|
|
-
Ознакомление с программой Muscle
Мной был получен файл с последовательностями дельта-антигенов в fasta-формате. (скачать) .
Полученный файл был импортирован в GeneDoc, где я попыталась вручную их выровнять: (скачать файл)
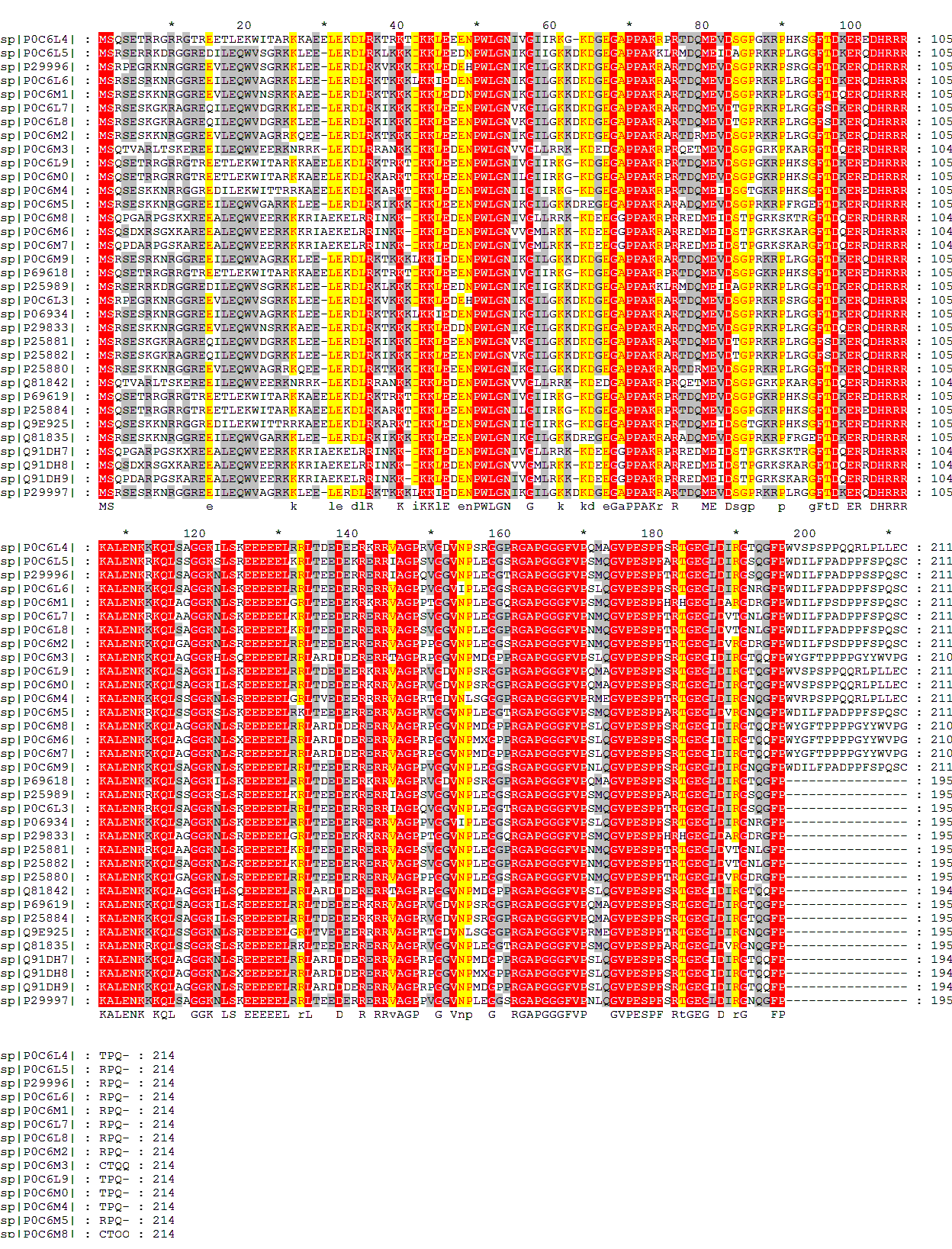 Далее, используя программу muscle, я получила множественное выравнивание этих последовательностей в fasta-формате: (скачать fasta-формате)
Далее, используя программу muscle, я получила множественное выравнивание этих последовательностей в fasta-формате: (скачать fasta-формате)
 В общем-то глядя на невыровненные последоваельности достаточно просто увидеть большую часть консервативных участков. В пользу этого сввидеетельсвует то, что участки со 103 по 196 столбцы выравнивания я и muscle выровняли одинаково. Да и остальная часть моего выравнивания достаточно похожа на предложенное muscle, хоть некоторые группы мне всё-таки не удалось заметить.
Это показывает преимущество множественного выравнивания перед парным: очевидность консервативных позиций.
В общем-то глядя на невыровненные последоваельности достаточно просто увидеть большую часть консервативных участков. В пользу этого сввидеетельсвует то, что участки со 103 по 196 столбцы выравнивания я и muscle выровняли одинаково. Да и остальная часть моего выравнивания достаточно похожа на предложенное muscle, хоть некоторые группы мне всё-таки не удалось заметить.
Это показывает преимущество множественного выравнивания перед парным: очевидность консервативных позиций.
-
Выравнивание набора гомологов своего белка
Для множественного выравнивания с моим белком с помощью BLASTp были выбраны следующие белки:
- IHFA_PHOLL E-value 2e-46, процент идентичности 90%
- IHFA_ALTMD E-value 9e-44, процент идентичности 84%
- IHFA_PASMU E-value 8e-35, процент идентичности 71%
- IHFA_NITMU E-value 9e-33, процент идентичности 68%
- IHFA_DICNV E-value 8e-31, процент идентичности 62%
- IHFA1_DECAR E-value 6e-27, процент идентичности 57%
- IHFA_POLSJ E-value 9e-25, процент идентичности 53%
Было получено следующее выравнивание: (скачать в fasta-формате), (скачать в msf-формате)
 Итак, посмотрим на полученное muscle множественное выравнивание….
Достаточно много консервативных остатков, но можно выделить такие протяженные участки:
48-67 позиции выравнивания (48-67 ак остатки моего белка) – 9 остатков из 20 консервативны, 70-91 позиции выравнивания (70-91 ак остатки белка) – 16 из 22 консервативны. Есть и еще консервативные позиции. Есть консервативная пара TK (14-15 позиция выравнивания и 14-15 ак моего белка).
Вообще возникают только сомнения насчет совпавших лейцинов в 48 и 97 позициях выравнивания (ак с теми же номерами в белке). Особенно последний случай, т.к. там вроде нет никакой «группировки» из консервативных остатков, а лейцин к тому же частая ак.
Итак, посмотрим на полученное muscle множественное выравнивание….
Достаточно много консервативных остатков, но можно выделить такие протяженные участки:
48-67 позиции выравнивания (48-67 ак остатки моего белка) – 9 остатков из 20 консервативны, 70-91 позиции выравнивания (70-91 ак остатки белка) – 16 из 22 консервативны. Есть и еще консервативные позиции. Есть консервативная пара TK (14-15 позиция выравнивания и 14-15 ак моего белка).
Вообще возникают только сомнения насчет совпавших лейцинов в 48 и 97 позициях выравнивания (ак с теми же номерами в белке). Особенно последний случай, т.к. там вроде нет никакой «группировки» из консервативных остатков, а лейцин к тому же частая ак.
-
Другие программы множественного выравнивания
Mafft
Теперь построим выравнивание для белков из предыдущего задания с помощью другой программы множественного выравнивания - mafft.
Выходной файл в fasta-формате импортируем в GeneDoc. (скачать выравнивание)
 сравним выравнивание muscle и mafft.
По сути, единственное, в чем они отличаются, это выравнивание начала последвательностей.
А именно в muscle мы уже видим в 11 позиции выравнивания, что у всех белков, кроме одного в этой позиции метионин.
Но, если посмотреть на начало этого выбившегося белка, то там тоже метионин. И если поставить в остальных белках гэп длиной 10 остатков в этом месте, что и делает mafft, то у нас появится консервативная позиция.
По-видимому, muscle не делает этого из-за других значений штрафов за гэпы. В остальном же эти выравнивания идентичны.
сравним выравнивание muscle и mafft.
По сути, единственное, в чем они отличаются, это выравнивание начала последвательностей.
А именно в muscle мы уже видим в 11 позиции выравнивания, что у всех белков, кроме одного в этой позиции метионин.
Но, если посмотреть на начало этого выбившегося белка, то там тоже метионин. И если поставить в остальных белках гэп длиной 10 остатков в этом месте, что и делает mafft, то у нас появится консервативная позиция.
По-видимому, muscle не делает этого из-за других значений штрафов за гэпы. В остальном же эти выравнивания идентичны.
Edialign
Выравнивание Edialign и Mafft полностью совпали, что опять же подтверждает важность найденной консервативной позиции. Думаю, следует сделать вывод, что иногда Mafft и Edialign чуть получше muscle.
-
Знакомство с некоторыми программами обработки множественных выравниваний
Consambig
Насколько я понимаю, программа на основе заданного выравниваиня по сути составляет паттерн (хоть и не в классической записи), под который подходят все заданные последовательности.
По выравниванию, полученному в предыдущем задании с помощью программы mafft, была составлена следующая последовательность: (скачать)
MefsvesletpXJTKXXXXXXJXXXXXXXXXXXXXXXXXXXXXXXXXLXXGXXXKJXGFG
XFXXRXKXXRPGRNPXTGEXXPXXARRVVXFXXXXXLXXXXXXXXxxXlkavXxX
Такая последовательность отлично показывает нам местонахождение консервативных и полуконсервативный позиций.
Distmat
Эта программа составляет матрицу эволюционных расстояний между всеми парами белков, приведенных в заданном выравнивании. по заданному выравниванию от mafft, получена следующая матрица: (скачать)
1 2 3 4 5 6 7 8
0.00 61.66 69.59 66.16 57.79 77.17 67.20 73.48
IHFA1_DECAR 1
0.00 56.15 16.97 45.73 39.28 17.17 68.49
IHFA_ALTMD 2
0.00 52.10 60.40 56.15 54.10 85.47
IHFA_DICNV 3
0.00 45.73 37.60 8.66 70.89
IHFA_ECOLI 4
0.00 48.23 39.28 61.99
IHFA_NITMU 5
0.00 39.28 85.47
IHFA_PASMU 6
0.00 69.59
IHFA_PHOLL 7
0.00
IHFA_POLSJ 8
Самое котороткое эволюционное расстояние мы наблюдаем между IHFA_ECOLI и IHFA_PHOLL, что подтверждает их большой процент идентичности и очень маленькое E-value.
Plotcon
Программа строит график, отражающий сходство ак остатков в различных позиуиях выравнивания. По тому же выравниванию получили следующий гафик (скачать)
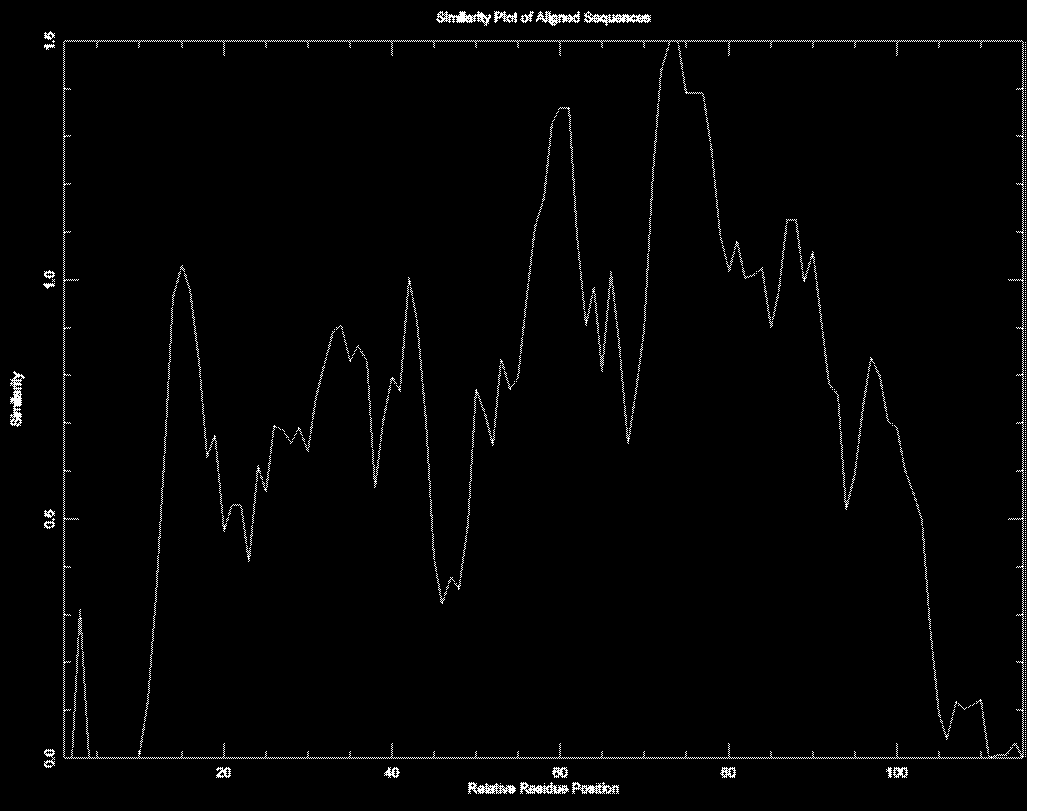 Как видно, график хорошо отражает окнсервативные участки. Особенно четко можно выделить два участка с наибольшим количеством пиков. примерно, с 55 по 65 и 70 по 95. Это примерно соответствует участкам с наибольшим числом консервативных остатков в этом выравниваии.
Как видно, график хорошо отражает окнсервативные участки. Особенно четко можно выделить два участка с наибольшим количеством пиков. примерно, с 55 по 65 и 70 по 95. Это примерно соответствует участкам с наибольшим числом консервативных остатков в этом выравниваии.
|