На страницу шестого семестра
Гомологичное моделирование комплекса белка с лигандом
Задание 1
Цель данного занятия ознакомится с возможностями гомологичного моделирования комплекса белка с лигандом. В этом занятии мы будем пользоваться пакетом Modeller.Работа будет производиться над белком из LYS_LAMBD(Enterobacteria phage lambda). Как образец берём известную структуру лизоцима форели. Задача - построение комплекса белка с лигандом.
MODELLER для моделирования структуры белков, в качестве входных данных нужны: управляющий скрипт, файл pdb со структурой-образцом, файл выравнивания с дополнительной информацией.
1) Построим выравнивание последовательности из структуры ID: 1lmp и лизоцима из LYS_LAMBD . Используем muscle. Полученное выравнивание сохраняем в формате PIR.
2) Модифицируем его:
3) Модификация файла со структуройПереименуем последовательность в файле выравнивания : Было >P1;uniprot|P*****|LYSC_LAMBD Стало >P1;seq Было >P1;1LMP|PDBID|CHAIN|SEQUENCE Стало >P1;1lmp После имени последовательности моделируемого белка добавляем строчку: sequence:ХХХХХ::::::: 0.00: 0.00 эта строчка описывает входные параметры последовательности для modeller. После имени последовательности белка-образца добавляем: structureX:1lmp_now.ent:1 :A: 132 :A:undefined:undefined:-1.00:-1.00 Эта строчка описывает, какой файл содержит структуру белка с этой последовательностью, номера первой и последней аминокислот в структуре, идентификатор цепи и т.д. В конце каждой последовательности добавьте символы /.
4) Создание управляющего скрипта lysc_lambd.py. Впишем возможные водородные связи с лигандом. В исходнике имеются взаимодействия следующих атомов:Удаляем всю воду из структуры (в текстовом редакторе), всем атомам лиганда присвоим один и тот же номер "остатка" (MODELLER считает, что один лиганд = один остаток) и модифицируем имена атомов каждого остатка, добавив в конец буквы A, B, C. Смысл операции в том, что атомы остатка 130 имели индекс А, атомы остатка 131 имели индекс В и т.д. После модификации имен атомов изменяем номера остатков на 130.
/1LMP//A/TRP`63/NE1 /1LMP//A/NAG`131/O3 /1LMP//A/NAG`131/O7 /1LMP//A/ASN`59/N /1LMP//A/ASP`52/OD2 /1LMP//A/NDG`132/O1L /1LMP//A/NDG`132/C1 /1LMP//A/NDG`132/O /1LMP//A/NDG`132/O1L /1LMP//A/ASN`46/OD1 /1LMP//A/VAL`109/N /1LMP//A/NDG`132/O6 /1LMP//A/GLU`35/OE2 /1LMP//A/NAG`131/N2 /1LMP//A/ALA`107/O /1LMP//A/NAG`130/O7 /1LMP//A/ASN`103/ND2 /1LMP//A/NAG`130/O6 /1LMP//A/ASP`101/OD2Cопоставляя соответствие остатков в выравнивании, с их принципиальной возможностью образовывать водородные связи, отметим атомы с которыми возможно взаимодейсвие в новой структуре
/1LMP//A/TRP`74/NE1 NAG`131/O3
/1LMP//A/LEU`70/N NAG`131/O7
/1LMP//A/GLN`136/O NAG`131/N2
/1LMP//A/PRO`57/N NDG`132/O1L
/1LMP//A/LYS`138/N NDG`132/O6
Получившийся скрипт:
from modeller.automodel import *
class mymodel(automodel):
def special_restraints(self, aln):
rsr = self.restraints
for ids in (('NE1:74:A', 'O3B:159:B'),
('N:70:A', 'O7B:159:B'),
('O:136:A', 'N2B:159:B'),
('N:138:A', 'O6C:159:B')):
atoms = [self.atoms[i] for i in ids]
rsr.add(forms.upper_bound(group=physical.upper_distance,
feature=features.distance(*atoms), mean=3.5, stdev=0.1))
env = environ()
env.io.hetatm = True
a = mymodel(env, alnfile='all_aligned.pir', knowns=('1lmp'), sequence='seq')
a.starting_model = 1
a.ending_model = 5
a.make()
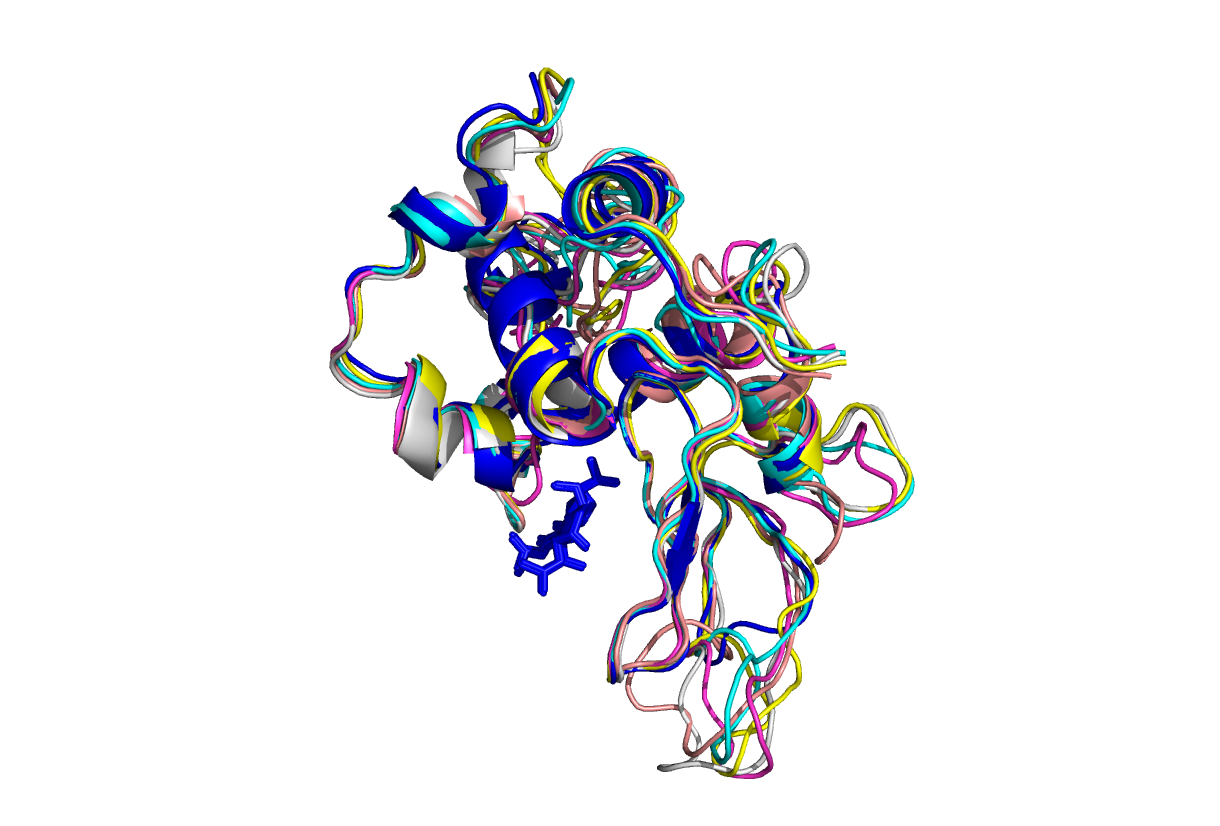
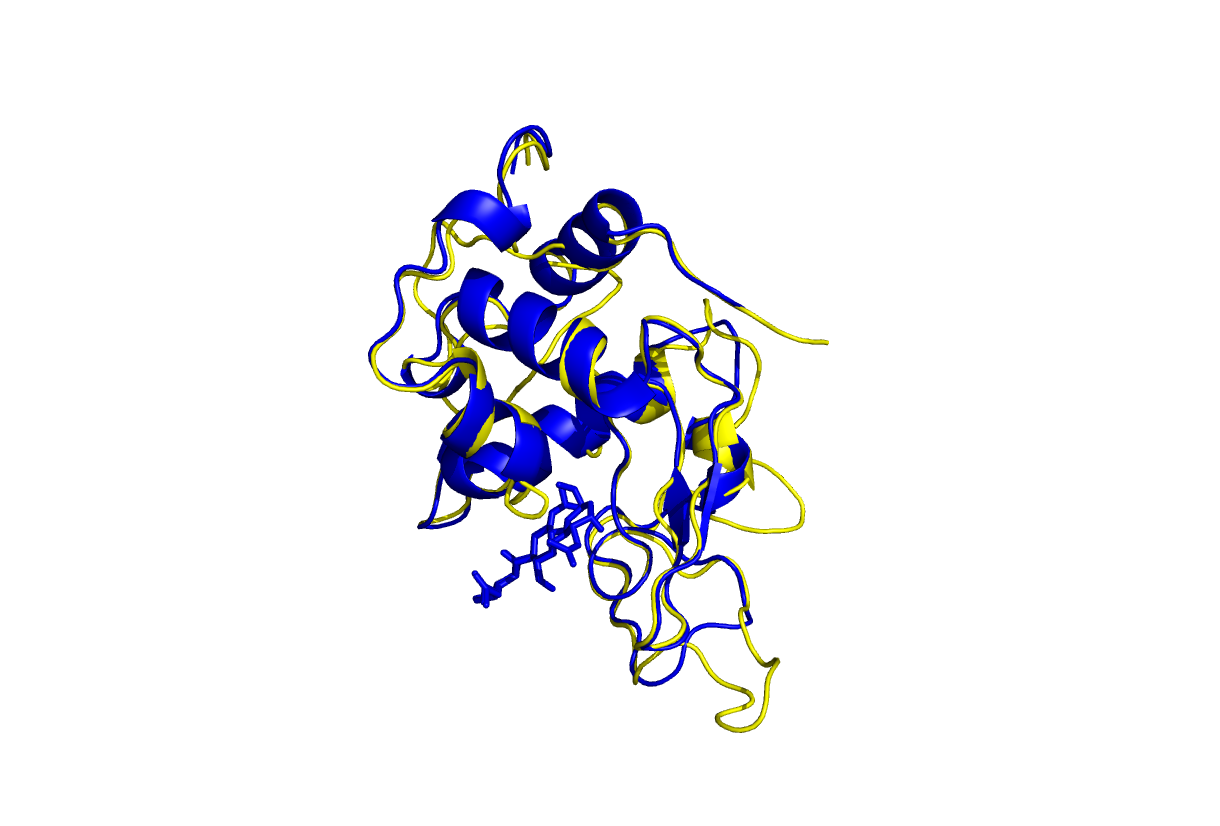
Получили пять моделей. На картинке синий полипептид - молекула лизоцима радужной форели.
Выбрали три критерия для оценки наилучшей модели.
| Модель | Anomalous bond lengths | Ramachandran | Omega |
| 1lmp |
Связи, длины которых отличаются от нормальных, с учётом дисперсии 130 NAG ( 130 ) A N2 C2 1.17 -6.2 131 NAG ( 131 ) A N2 C2 1.13 -7.1 RMS Z-score for bond lengths: 0.839 RMS-deviation in bond distances: 0.023 Variance: 94.771 (Under-)estimated Z-score: 7.175 |
Z-score : -0.965 | Omega average and std. deviation= 179.870 2.718 Significant deviations from expected 5.5!! |
| 1 |
137 HIS ( 137 ) A CA C 1.42 -5.2 138 LYS ( 138 ) A N CA 1.35 -5.6 RMS Z-score for bond lengths: 1.064 RMS-deviation in bond distances: 0.021 |
Z-score : -5.013 | Omega average and std. deviation= 180.553 5.924(agrees) |
| 2 |
RMS Z-score for bond lengths: 1.078 RMS-deviation in bond distances: 0.021 |
Z-score : -5.095 | Omega average and std. deviation= 182.107 7.036 Significant deviations from expected 5.5!!! |
| 3 |
137 HIS ( 137 ) A CA C 1.41 -5.4 138 LYS ( 138 ) A N CA 1.35 -5.8 RMS Z-score for bond lengths: 1.066 RMS-deviation in bond distances: 0.021 |
Z-score : -3.427 | Omega average and std. deviation= 180.725 6.445(agrees) |
| 4 |
137 HIS ( 137 ) A CA C 1.43 -4.4 138 LYS ( 138 ) A N CA 1.36 -5.3 RMS Z-score for bond lengths: 1.057 RMS-deviation in bond distances: 0.021 |
Z-score : -4.596 | Omega average and std. deviation= 181.515 6.742(agrees) |
| 5 |
124 TRP ( 124 ) A CA C 1.44 -4.2 137 HIS ( 137 ) A CA C 1.42 -5.1 138 LYS ( 138 ) A N CA 1.35 -5.5 146 PHE ( 146 ) A CB CG 1.64 5.9 RMS Z-score for bond lengths: 1.093 RMS-deviation in bond distances: 0.022 |
Z-score : -4.657 | Omega average and std. deviation= 181.319 6.297(agrees) |
Думаю, что лучше всего по критериям проходит третья модель. На картинке - жёлтая.