На страницу третьего семестра
Комплексы ДНК-белок
Поиск ДНК-белковых контактов в заданной структуре
Упр.1
Скрипт к упражнению 1 (определение множеств атомов). script.spt
Упр.2
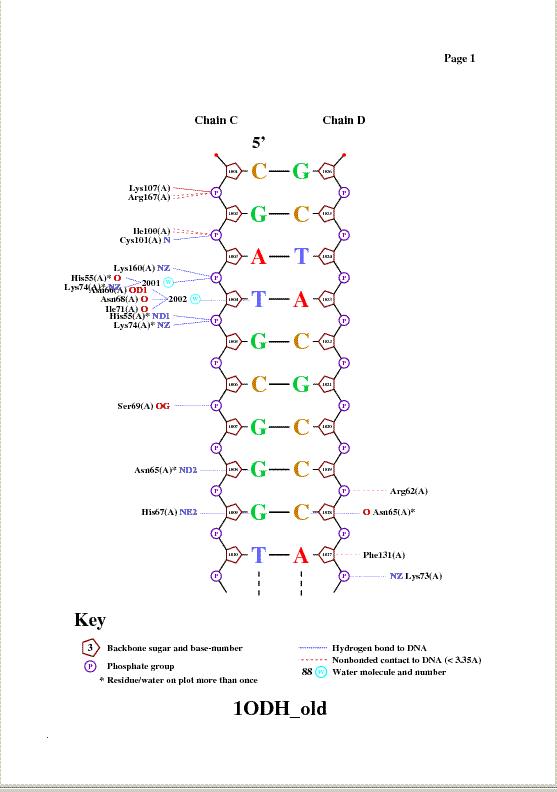
Таблица. Контакты разного типа в комплексе 1ODH.pdb
Контакты атомов белка с Полярные Неполярные Всего остатками 2'-дезоксирибозы 8 9 17 остатками фосфорной кислоты 19 8 27 остатками азотистых оснований со стороны большой бороздки 3 нет 3 остатками азотистых оснований со стороны малой бороздки нет нет нет Больше полярных контактов, отсутствуют неполярные контакты с остатками азотистых оснований, нет контактов со стороны малой бороздки.
Упр.3
Получение популярной схемы ДНК-белковых контактов с помощью программы nucplot
Предсказание вторичной структуры заданной тРНК
Упр.1. Предсказание вторичной структуры тРНК путем поиска инвертированных повторов
Для работы с программой einverted из текста pdb-файла была получена При запуске программы einverted нам задавались вопросы. При этом мы поменяли только один из параметров - "Minimum score threshold", его пришлось сделать очень маленьким (5 вместо 50), только в таком случае выдавались какие-то результаты ( Участки очень короткие, веса маленькие, поэтому и порог низкий) и сохранена в fasta-формате последовательность аспартил-тРНК.
Программа einverted способна показать нам только какой-то участок полной спирали, и то не всегда. Результатов поиска инвертированных повторов явно недостаточно для того, чтобы охарактеризовать вторичную структуру РНК, ведь она намного сложнее, чем совокупность таких повторов, образующих дуплексы, и одноцепочечных участков.
Упр.2. Предсказание вторичной структуры тРНК по алгоритму Зукера.
Для работы использовали такую команду: mfold SEQ=tRNA.fasta P=X где X - значение параметра P - число, означающее, на сколько процентов выдаваемое предсказание вторичной структуры может отличаться по своей вычисленной энергии от оптимального. В выдачах mfold необходимые нам файлы с расширением GIF, демонстрирующие изображение предсказанной структуры. Понятно, что чем больше значение P, тем больше степеней свободы и, соответственно, количество выдаваемых структур. Наиболее удовлетворительной показалась третья по счету структура, полученная при втором обращении к mfold (со значением P, равным 15-ти). Все, кроме выбранной, либо не похожи на клеверный лист вовсе, либо не обладают одним из стеблей.
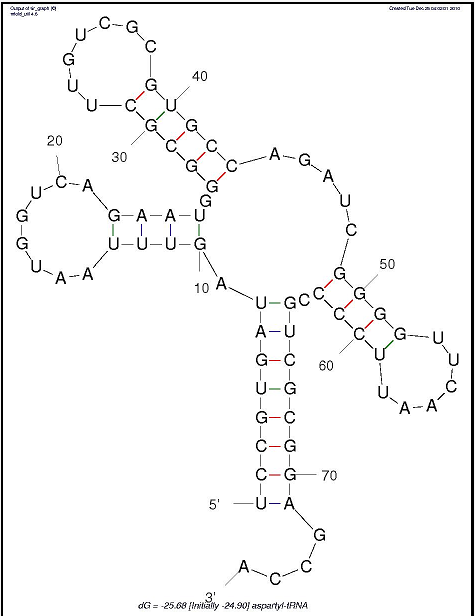
Участок структуры Позиции в структуре
(по результатам find_pair)Результаты предсказания
с помощью einvertedРезультаты предсказания
по алгоритму ЗукераАкцепторный стебель 5' 901-907 3'
5' 972-960 3'
Всего 7 парпредсказано 5 пар из 7 реальных предсказано 8 пар D-стебель 5' 938-944 3'
5' 932-926 3'
Всего 7 парпредсказано 0 пар предсказано 4 пары T-стебель 5' 949-954 3'
5' 965-958 3'
Всего 6 парпредсказано 0 пар предсказано 4 пары Антикодоновый стебель 5' 910-913 3'
5' 925-922 3'
Всего 3 парыпредсказано 0 пар предсказано 5 пар Общее число канонических пар нуклеотидов 23 5 21
Упр.4
На полученной схеме а) аминокислотный остаток с наибольшим числом указанных на схеме контактов с ДНК -Lys74(A) и His55(A), по два контакта.
б) аминокислотный остаток, по-моему мнению, наиболее важный для распознавания последовательности ДНК- Asn65, потому что во-первых взаимодействие идёт с остатком азотистого основания- гуанина, и оно само полярно и довольно специфично.
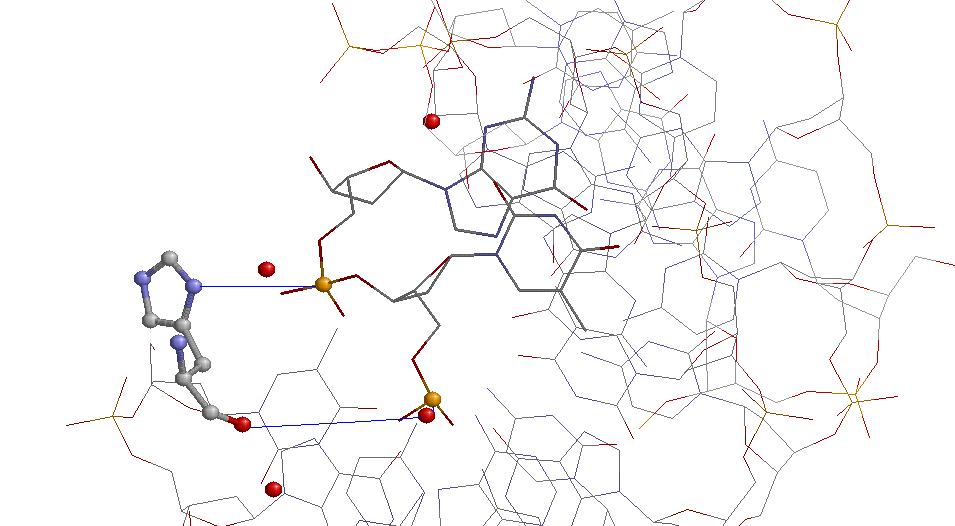
На изображении His55, взаимодействие идёт с двумя фосфорными остатками ДНК, в одном случае оно, видимо, идёт через кислород воды.
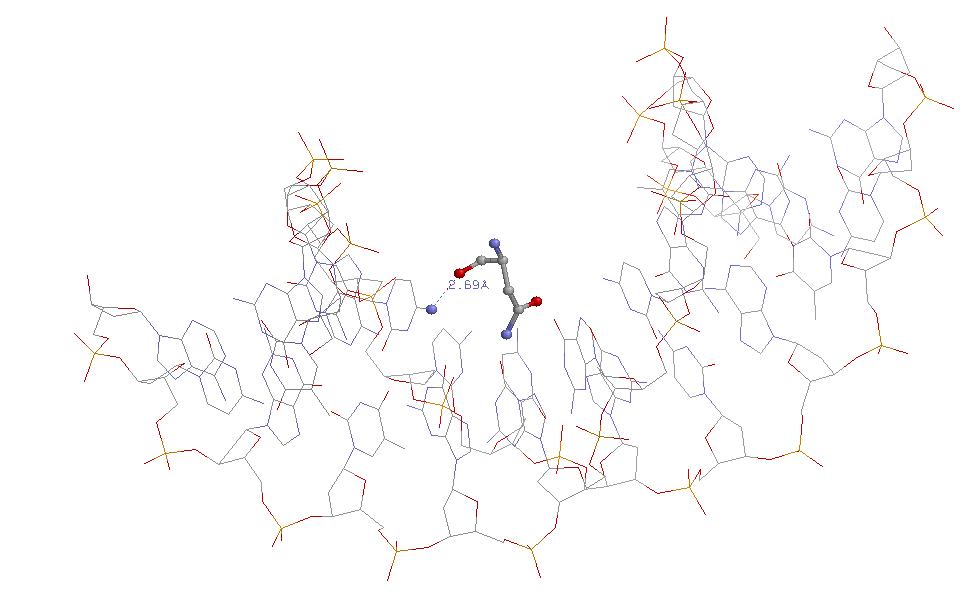
Аминокислотный остаток лизина, ДНК в остовной модели, взаимодействие.