|
|
|
|
|
|
|
|
|
|
|
 |
|
||||||||
|
|
|
|
|
|
|
|
|
||
Гомологичное моделирование комплекса белка с лигандомМы будем работать с белком лизоцимом
из организма курицы. Используя
известную структуру лизоцима форели
как образец, Построим выравнивание последовательности из структуры: LYSC_ONCMY и LYSC_CHICK с помощью Clustal.Выравнивание сохраните в формате PIR. Теперь переименуем последовательность в файле выравнивания(например) : >P1;1LMP__|PDBID|CHAIN|SEQUENCE заменим на >P1;1lmp После имени последовательности моделируемого белка добавим строчку: sequence:ХХХХХ::::::: 0.00: 0.00эта строчка описывает входные параметры последовательности для modeller. После имени последовательности белка-образца добавим: structureX:1lmp_now.ent:1 :A: 132 :A:undefined:undefined:-1.00:-1.00 эта строчка описывает, какой файл содержит структуру белка с этой последовательностью, номера первой и последней аминокислот в структуре, идентификатор цепи и т.д. В конце каждой последовательности добавим символы /. Символ "/" означает конец цепи белка. Точка указывает на то, что имеется один лиганд (если бы было два лиганда стояли бы две точки). Модификацируем файл со структурой:
удалим всю воду из структуры (в
текстовом редакторе)
всем атомам лиганда присвоим один и тот же номер "остатка" (MODELLER считает, что один лиганд = один остаток) в нашем случае это номер 130 и модифицируем имена атомов каждого остатка, добавив в конец буквы A, B, C.
Смысл операции в том, что атомы остатка 130 имели индекс А, атомы остатка 131 имели индекс В и т.д. . После модификации имен атомов изменим номера остатков на 130. HETATM 1014 O7 NAG 130 -> HETATM 1014 O7A NAG 130 сохраним в файле 1lmp_now.ent Созданим скрипт lysc_chick.py
from modeller.automodel import *
class mymodel(automodel):
def special_restraints(self, aln):
rsr = self.restraints
for ids in (('O:125:A', 'N2B:148:B'),
('ND2:121:A', 'O7A:148:B'),
('N:77:A', 'O7B:148:B')):
atoms = [self.atoms[i] for i in ids]
rsr.add(forms.upper_bound(group=physical.upper_distance,
feature=features.distance(*atoms), mean=3.5, stdev=0.1))
env = environ()
env.io.hetatm = True
a = mymodel(env, alnfile='align.pir', knowns=('1lmp'), sequence='seq')
a.starting_model = 1
a.ending_model = 5
a.make()
В скрипте указано:
Запустим исполнение скрипта командой mod9v7 lysc_chick.py & Получили 5 моделей:
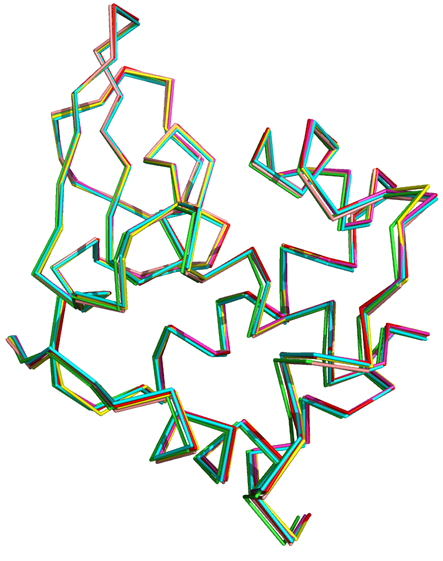
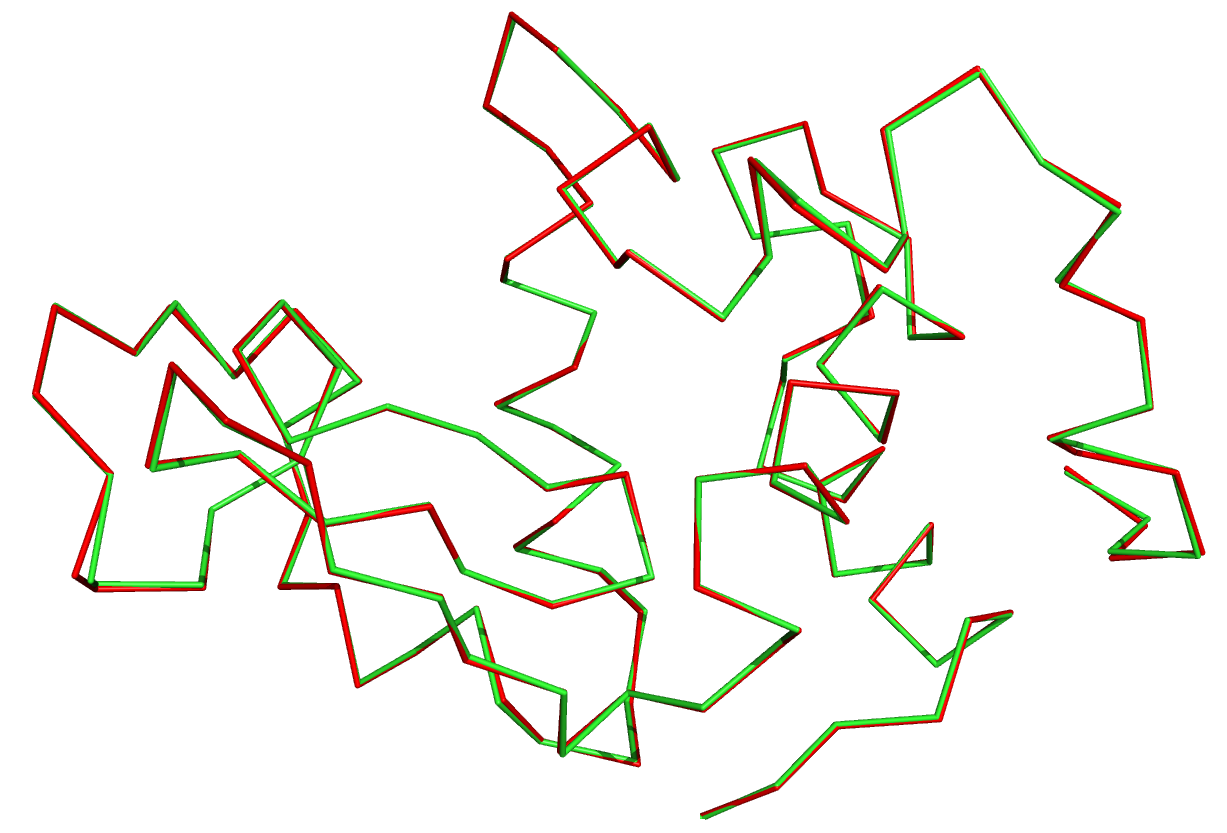
Все полученные модели очень близки друг к другу, также с каждой из моделей было сделано пространственное выравнивание с 1lmp и RMSD во всех случаях не превышало 0,2, что говорит о хорошем пространственном соотвествии. На данной картине красным отмечен белок 1lmp. Проверим качество моделей и выберите лучшую. Инструменты для оценки качества структуры можно найти в веб интерфейсе WHATIF. Достаточно 2-3 инструментов. 1 модель Structure Z-scores, positive is better than average: 2 модель Structure Z-scores, positive is better than average: 1st generation packing quality : -3.134 2nd generation packing quality : -2.433 Ramachandran plot appearance : -0.028 chi-1/chi-2 rotamer normality : -2.052 Backbone conformation : -0.571 RMS Z-scores, should be close to 1.0: Bond lengths : 0.927 Bond angles : 1.301 Omega angle restraints : 0.809 Side chain planarity : 0.478 (tight) Improper dihedral distribution : 1.025 Inside/Outside distribution : 1.083 3 модель Structure Z-scores, positive is better than average: 1st generation packing quality : -3.139 2nd generation packing quality : -2.820 Ramachandran plot appearance : 0.037 chi-1/chi-2 rotamer normality : -1.028 Backbone conformation : -0.484 RMS Z-scores, should be close to 1.0: Bond lengths : 0.922 Bond angles : 1.286 Omega angle restraints : 0.902 Side chain planarity : 0.369 (tight) Improper dihedral distribution : 1.054 Inside/Outside distribution : 1.079 4 модель Structure Z-scores, positive is better than average: 1st generation packing quality : -3.072 2nd generation packing quality : -2.516 Ramachandran plot appearance : 0.006 chi-1/chi-2 rotamer normality : -1.868 Backbone conformation : -0.317 RMS Z-scores, should be close to 1.0: Bond lengths : 0.920 Bond angles : 1.264 Omega angle restraints : 0.825 Side chain planarity : 0.273 (tight) Improper dihedral distribution : 1.041 Inside/Outside distribution : 1.072 5 модель Structure Z-scores, positive is better than average: 1st generation packing quality : -2.857 2nd generation packing quality : -2.471 Ramachandran plot appearance : 0.091 chi-1/chi-2 rotamer normality : -2.946 Backbone conformation : -0.175 RMS Z-scores, should be close to 1.0: Bond lengths : 0.925 Bond angles : 1.268 Omega angle restraints : 0.730 Side chain planarity : 0.347 (tight) Improper dihedral distribution : 1.027 Inside/Outside distribution : 1.085 Рассмотрим такие параметры как Ramachandran plot appearance(должна быть как можно больше) и Bond lengths(должна -> 1). Исходя из этого первая модель является лучшей.
RMSD = 0,196
|
| ||||||||
|
|
|||||||||
|
© Замараев Алексей |
|
||||||||