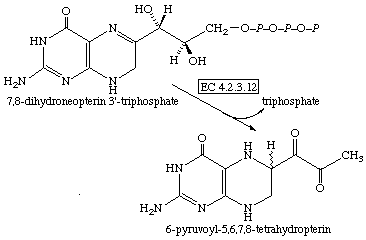
С <=> А + В
|
|
UniProt ID |
UniProt AC |
Имя гена |
Длина последовательности. |
Домен |
||
Идентификатор Pfam |
Положение в последовательности |
|||||
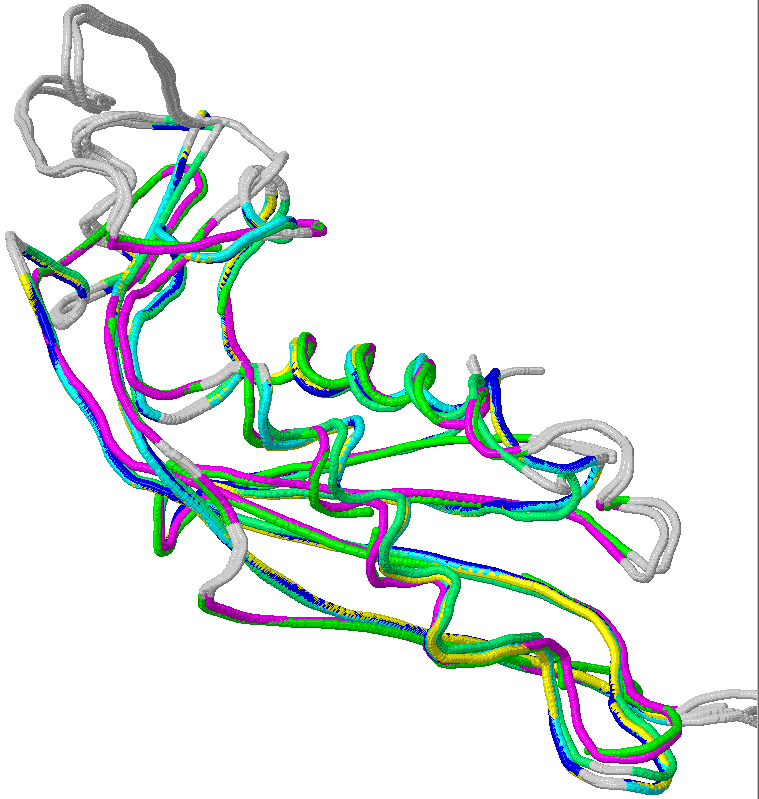
| 1 | PTPS_ECOLI | P65870, Q2MA64, Q46903 | 121 | YGCM | PF01242 | 1-121 |
| 2 | PTPS_HUMAN | Q03393, Q8WVG8 | PTS | 145 | PF01242 | 3-145 |

|
|
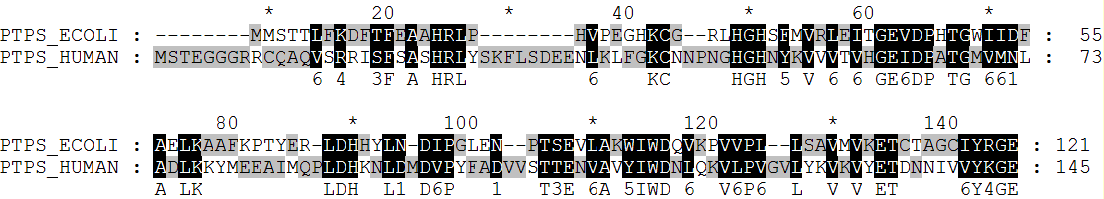
needle seq1.fasta seq2_hum.fasta
water seq1.fasta seq2_hum.fasta
#=======================================
#
# Aligned_sequences: 2
# 1: PTPS_ECOLI
# 2: PTPS_HUMAN
# Matrix: EBLOSUM62
# Gap_penalty: 10.0
# Extend_penalty: 0.5
#
# Length: 150
# Identity (ID): 47/150 (31.3%)
# Similarity: 75/150 (50.0%)
# Gaps: 34/150 (22.7%)
# Score: 153.5
#
#
#=======================================
PTPS_ECOLI 1 MMST---------TLFKDFTFEAAHRL--------PHVPEGHKCGRL--H 31
||| .:.:..:|.|:||| .::....||... |
PTPS_HUMAN 1 -MSTEGGGRRCQAQVSRRISFSASHRLYSKFLSDEENLKLFGKCNNPNGH 49
PTPS_ECOLI 32 GHSFMVRLEITGEVDPHTGWIIDFAELK-----AAFKPTYERLDHHYLN- 75
||::.|.:.:.||:||.||.:::.|:|| |..:| |||..|:
PTPS_HUMAN 50 GHNYKVVVTVHGEIDPATGMVMNLADLKKYMEEAIMQP----LDHKNLDM 95
PTPS_ECOLI 76 DIPGLEN--PTSEVLAKWIWDQVKPVVP--LLSAVMVKETCTAGCIYRGE 121
|:|...: .|:|.:|.:|||.::.|:| :|..|.|.||.....:|:||
PTPS_HUMAN 96 DVPYFADVVSTTENVAVYIWDNLQKVLPVGVLYKVKVYETDNNIVVYKGE 145
#---------------------------------------
#---------------------------------------
|
#=======================================
#
# Aligned_sequences: 2
# 1: PTPS_ECOLI
# 2: PTPS_HUMAN
# Matrix: EBLOSUM62
# Gap_penalty: 10.0
# Extend_penalty: 0.5
#
# Length: 131
# Identity: 44/131 (33.6%)
# Similarity: 70/131 (53.4%)
# Gaps: 24/131 (18.3%)
# Score: 155.5
#
#
#=======================================
PTPS_ECOLI 11 TFEAAHRL--------PHVPEGHKCGRL--HGHSFMVRLEITGEVDPHTG 50
:|.|:||| .::....||... |||::.|.:.:.||:||.||
PTPS_HUMAN 19 SFSASHRLYSKFLSDEENLKLFGKCNNPNGHGHNYKVVVTVHGEIDPATG 68
PTPS_ECOLI 51 WIIDFAELK-----AAFKPTYERLDHHYLN-DIPGLEN--PTSEVLAKWI 92
.:::.|:|| |..:| |||..|: |:|...: .|:|.:|.:|
PTPS_HUMAN 69 MVMNLADLKKYMEEAIMQP----LDHKNLDMDVPYFADVVSTTENVAVYI 114
PTPS_ECOLI 93 WDQVKPVVP--LLSAVMVKETCTAGCIYRGE 121
||.::.|:| :|..|.|.||.....:|:||
PTPS_HUMAN 115 WDNLQKVLPVGVLYKVKVYETDNNIVVYKGE 145
#---------------------------------------
#---------------------------------------
|
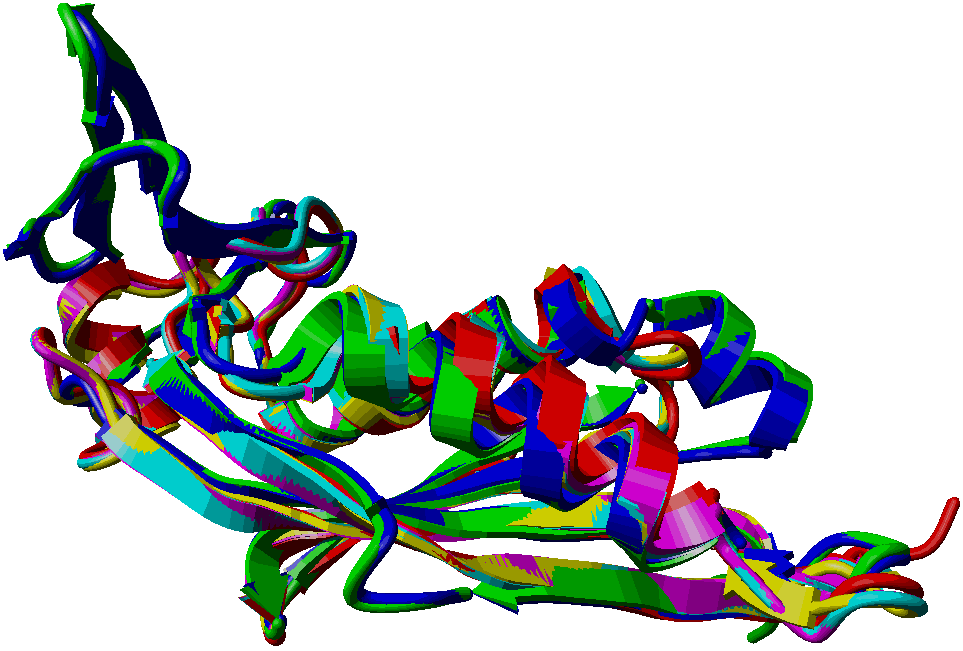
|
|

|

|