| (А:120,((((B:10,C:10):50,D:65):5,E:70):5,F:70):30); |
- Изображение дерева, описанного этой формулой:
 |
Данное дерево - укоренённое неультраметрическое.
- Описание ветвей дерева как разбиения множества листьев
| A | B | C | D | E | F |
| - | - | - | * | - | - |
| - | * | * | - | - | - |
| - | * | * | * | - | - |
| - | - | - | - | * | - |
| * | - | - | - | - | * |
| - | - | - | - | - | * |
| * | - | - | - | - | - |
| - | * | - | - | - | - |
| - | - | * | - | - | - |
Поэтому, окончательный вариант записи состоит всего из трёх строк:
| A | B | C | D | E | F |
| - | * | * | - | - | - |
| - | * | * | * | - | - |
| * | - | - | - | - | * |
- Получение искуственных мутантных последовательностей
| 645 * R/ 100 |
Для того, чтобы воспользоваться программой msbar пакета EMBOSS, был написан скрипт, строки которого имеют вид:
msbar infile outfile -point 4 -count n -auto |
Файл сохранён как текст UNIX.
- Реконструкция дерева алгоритмами UPGMA, Neighbor-joining и максимального правдоподобия.
- Алгоритм максимального правдоподобия
fdnaml offsprings.fasta -ttratio 1 -auto |
+------------------F |
- Алгоритм Neighbor-joining
fdnadist offsprings.fasta -ttratio 1 -auto |
fneighbor offsprings.fdnadist -auto |
+--B +---------1 ! +-C ! ! +----------------D 2--3 ! ! +------------------E ! +-4 ! +------------------F ! +-------------------------------------A |
- Алгоритм UPGMA
fneighbor offsprings.fdnadist -treetype u -auto |
В результате получилось изображение:
+------------------------------------------------------A ! ! +---B ! +-------------------------1 --5 +---2 +---C ! ! ! ! +--3 +-----------------------------D ! ! ! +-----------------4 +---------------------------------E ! +------------------------------------F |
- Анализ полученных данных
| Алгоритм | Рисунок дерева в бескорневом виде | Описание ветвей дерева как разбиения множества листьев |
| Исходное дерево | 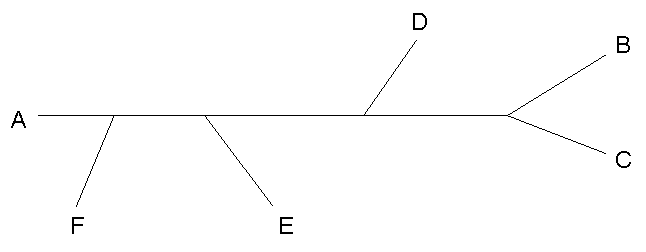 |
ABCDEF -**--- -***-- *----* |
| Максимального правдоподобия |  |
ABCDEF -**--- ---*** ----** |
| Neighbor-joining |  |
ABCDEF -**--- ---*** ----** |
| UPGMA |  |
ABCDEF -**--- -***-- *----* |
Итак, по таблице видно, что наиболее похожий, а вернее точный, результат выдал четвёртый рассмотренный алгоритм - UPGMA. Однако, если исходные данные таковы, что гипотеза молекулярных часов не проходит, то реконструкция укоренённого дерева, как в алгоритме UPGMA, намного менее надёжна, чем реконструкция неукоренённого.
Дополнительное задание
Сравнение разных способов оценки эволюционных расстояний между генами.
Основная задача — определить границы применимости 3-х способов оценки эволюционного расстояния между нуклеотидными последовательностями.- Определить "истинные" эволюционные расстояния между конечными мутантами в cозданной модели эволюции (т.е. расстояния в числе замен между листьями заданного дерева)
На стр. "All_data" размещена таблица с "истинными" попарными расстояниями (число замен на 100 нуклеотидов):
| узел | A | B | C | D | E | F | P | T | G | H |
| A | 0 | 220 | 220 | 225 | 225 | 220 | 210 | 160 | 155 | 150 |
| B | 0 | 20 | 125 | 135 | 140 | 10 | 60 | 65 | 70 | |
| C | 0 | 125 | 135 | 140 | 10 | 60 | 65 | 70 | ||
| D | 0 | 140 | 145 | 115 | 65 | 70 | 75 | |||
| E | 0 | 145 | 125 | 75 | 70 | 75 | ||||
| F | 0 | 130 | 80 | 75 | 70 | |||||
| P | 0 | 50 | 55 | 60 | ||||||
| T | 0 | 5 | 10 | |||||||
| G | 0 | 5 | ||||||||
| H | 0 |
- Определить попарные эволюционные расстояния между всеми последовательностями (включая исходную) c помощью программы distmat пакета EMBOSS.
Методы оценки эволюционных расстояний программой distmat:
0 : Uncorrected
1 : Jukes-Cantor
2 : Kimura
3 : Tamura
4 : Tajima-Nei
5 : Jin-Nei Gamma
Выбирая соответствующие пункты меню программы, получены 3 матрицы попарных расстояний:
- матрица попарных различий (D) (uncorrected distances);
distmat -sequence offsprings.fasta -outfile offsprings.distmat -nucmethod 0 |
1 2 3 4 5 6 |
- матрица попарных расстояний, вычисленных по формуле Джукса - Кантора (JC)
distmat -sequence offsprings.fasta -outfile offsprings.distmat -nucmethod 1 |
1 2 3 4 5 6 |
- матрица попарных расстояний, вычисленных по модели М.Кимура.
distmat -sequence offsprings.fasta -outfile offsprings.distmat -nucmethod 2 |
1 2 3 4 5 6 |
Матрица перенесена на лист "All_Data".
- Сравнение методов оценки эволюционных расстояний, предложеннных программой distmat
| "истинные" попарные расстояния | Матрица попарного различия | метод Джукса – Кантора | метод М.Кимура | |
| AD | 225 | 67,44 | 172,11 | 172,41 |
| AE | 225 | 68,06 | 178,54 | 178,55 |
| AB | 220 | 65,89 | 158,12 | 161,1 |
| AC | 220 | 65,27 | 153,18 | 156,23 |
| AF | 220 | 67,44 | 172,11 | 172,94 |
| DF | 145 | 57,52 | 109,23 | 109,25 |
| EF | 145 | 58,29 | 112,63 | 112,78 |
| BF | 140 | 58,76 | 114,75 | 114,75 |
| CF | 140 | 58,91 | 115,47 | 115,51 |
| DE | 140 | 56,59 | 105,34 | 106,77 |
| BE | 135 | 56,59 | 105,34 | 105,52 |
| CE | 135 | 54,73 | 98,12 | 98,2 |
| BD | 125 | 53,18 | 92,59 | 92,6 |
| CD | 125 | 53,18 | 92,59 | 92,68 |
| BC | 20 | 12,4 | 13,56 | 13,56 |

Из графика видно, что кривые, соответствующие методам Джукса-Кантора и М.Кимура практически полностью совпадают. Для того, чтобы различия между методами были более заметны, построен график, на котором по оси абсцисс отложен условный номер пары расстояний, а по оси ординат - нормированные значения чисел мутаций (см лист "Comparison_2"). По совпадению с кривой "истинных" значений можно судить о точности метода.

По графику можно заметить, что хоть кривые, отображающие методы Джукса-Кантора и М.Кимура, почти всё время совпадают, зелёная кривая всё же выше малиновой, а, значит, оценки эволюционных расстояний, полученные методом М.Кимура ближе к "истинным", чем, полученные методом Джукса-Кантора. Такой результат объясняется отличием этих моделей*: в модели Джукса - Кантора транзиции и трансверсии считаются равновероятными, а в модели М.Кимура - нет (транзиции более вероятны). В связи с этим получается, что метод оценки эволюционного расстояния между нуклеотидными последовательностями М.Кимура более верен, потому что в его основе лежит более точная модель. И хотя на данном примере различия между этими методами минимальны, при увеличении числа мутаций - эволюционного расстояния, погрешность метода Джукса - Кантора будет накапливаться и, в силу этого, данный метод будет неприменим.
Метод попарных различий показал наихудший результат - его кривая почти параллельна оси абсцисс и совпадает с кривой "истинных" значений почти только в 1 точке.
Интересно отметить, что с увеличением номера пары все три метода стремятся в одну точку и при этом почти совпадают с "истинным" значением. Это объясняется тем, что в силу сортировки по "истинным" расстояниям первые рассмотренные пары находятся далеко друг от друга (см. исходное дерево и сравнительную таблицу), а последующие пары рассматривались с меньшим расстоянием. Отсюда делаем вывод, что чем меньше эволюционное расстояние между потомками, тем более точны все 3 рассмотренных метода, однако при увеличении эволюционнного расстояния они сильно расходятся как друг от друга, так и от "истинной" картины. Иными словами, получили такую картинку, только отражённую относительно оси ординат:

Далее в таблице предложены диапозоны значений эволюционных расстояний для каждого из изученных методов:
| Способ оценки эволюционных расстояний | Диапозон близких** к "истинному" значений | Диапозон максимально отличающихся*** от "истинного" значений |
| Метод попарных различий | до 20-25 замен на 100 нуклеотидов | от 125 замен на 100 нуклеотидов |
| Метод Джукса-Кантора | до 20-25 замен на 100 нуклеотидов | от 220 замен на 100 нуклеотидов |
| Метод М.Кимура | до 20-25 замен на 100 нуклеотидов | от 222 замен на 100 нуклеотидов |
Значит, для очень "дальних родственников" лучше использовать метод М.Кимура, для более близких последовательностей - Джукса - Кантора, а для очень близких - матрицу попарных различий, поскольку этот метод не требует особых компьютерных ресурсов.
* Сходство моделей Джукса-Кантора и М.Кимура:
| Модель Джукса-Кантора | Модель М.Кимура |
Расстояние = -b ln (1-D/b) |
Расстояние = -0.5 ln[ (1-2P-Q)*sqrt(1-2Q)] |
Расстояние = -const ln [(1-T)*P]
где const<1, P<1..
Значит, при небольшом числе рассматриваемых мутаций, заключающихся в заменах нуклеотидов, значительного различия между моделями наблюдаться не будет, так как основное отличие в значениях получается за счёт того, что в модели М.Кимура возможности транверсий и транзиций не равновероятны.
** Близкими к "истинным" считаются значения, при которых примые на второй диаграмме максимально совпадают. Как видно из рисунка, это наблюдается в районе 15 пары. Далее, в соответствии с таблицей, а также excel-файла, было отмечено, что 15-ой паре соответствует 20 замен, но, поскольку, совпадение прямых ещё немного продолжается левее, диапозон был выбран 20-25 замен на 100 нуклеотидов (что лучше заметно в исходном файле, особенно при вариации цены делений и разметки, которой нет на диаграмме, чтобы не загромождать рисунок).
*** Максимально отличающимися от "истинных" считаются значения, при достижении которых кривая, полученная определённым методом начинает максимально отдаляться от кривой "истинных" значений. Для метода попарных различий это происходит примерно на уровне 14 пары, отсюда и диапозон: от 125 замен на 100 нуклеотидов; для метода Джукса-Кантора - на уровне 4 пары, значит, от 220 замен на 100 нуклеотидов; для метода М.Кимура это тоже происходит на уровне 4 пары, но, поскольку зелёная кривая всё же немного выше мальновой, диапозон выбран от 222 замен на 100 нуклеотидов. Аналогично, нагляднее диапозоны видны в исходном файле.
Значит, при небольшом числе рассматриваемых мутаций, заключающихся в заменах нуклеотидов, значительного различия между моделями наблюдаться не будет, так как основное отличие в значениях получается за счёт того, что в модели М.Кимура возможности транверсий и транзиций не равновероятны.
** Близкими к "истинным" считаются значения, при которых примые на второй диаграмме максимально совпадают. Как видно из рисунка, это наблюдается в районе 15 пары. Далее, в соответствии с таблицей, а также excel-файла, было отмечено, что 15-ой паре соответствует 20 замен, но, поскольку, совпадение прямых ещё немного продолжается левее, диапозон был выбран 20-25 замен на 100 нуклеотидов (что лучше заметно в исходном файле, особенно при вариации цены делений и разметки, которой нет на диаграмме, чтобы не загромождать рисунок).
*** Максимально отличающимися от "истинных" считаются значения, при достижении которых кривая, полученная определённым методом начинает максимально отдаляться от кривой "истинных" значений. Для метода попарных различий это происходит примерно на уровне 14 пары, отсюда и диапозон: от 125 замен на 100 нуклеотидов; для метода Джукса-Кантора - на уровне 4 пары, значит, от 220 замен на 100 нуклеотидов; для метода М.Кимура это тоже происходит на уровне 4 пары, но, поскольку зелёная кривая всё же немного выше мальновой, диапозон выбран от 222 замен на 100 нуклеотидов. Аналогично, нагляднее диапозоны видны в исходном файле.
Главная Первый семестр Второй семестр Третий семестр