Вам даны файлы:
-
файл праметров для минимизации энергии em.mdp.
-
файл праметров для "утряски" воды pr.mdp pr.mdp.
-
файл праметров для молекулярной динамики md.mdp.
С помощью программы fiber из пакета 3DNA постройте небольшой дуплекс с последовательностью GATCTA. Для того, что бы fiber работал надо задать путь и переменную среды:
export X3DNA=/home/preps/golovin/progs/X3DNA
export PATH=/home/preps/golovin/progs/X3DNA/bin:${PATH}
Построим файл топологии системы в силовом поле amber99sb и файл с координатами в формате Gromacs. Предполагается, что структура дуплекса находится в файле dna.pdb.
pdb2gmx -f dna.pdb -o dna -p dna -ff amber99sb -water tip3p
Удалим 5' фосфаты из структуры дуплекса. Их два на 5' конце кажой цепи. Также надо изменить имена нуклеотидов добвавив D к названию нуклеотида," Т"->"DT" (пример для vim: :%s/ \([GATC]\) \([AB]\)/ D\1 \2/). И заменить имя атома "С5М" на " С7".
Сделаем небольшой отступ в ячейке от ДНК
editconf -f dna.gro -o dna_ec -d 1.5
Проведём оптимизацию геометрии системы, что бы удалить "плохие" контакты в молекуле.
grompp -f em -c dna_ec -p dna -o dna_em -maxwarn 1
mdrun -deffnm dna_em -v
Изменение максимальной силы в ходе оптимизации геометрии:
начальное значение максимальной силы = Step 0 Fmax = 4.789e+03,
конечное значение максимальной силы = Step 54 Fmax = 9.566e+02.
Добавим в ячейку молекулы воды:
genbox -cp dna_em -p dna -cs -o dna_s
Нейтрализуем заряд системы. Это делаем в два шага: строим tpr и запускаем genion:
grompp -f em -p dna -c dna_s -o dna_s
genion -s dna_s -o dna_si -p dna -np 10
Количество положительных ионов необходимых для нейтрализации заряда системы равно 10, тк заряд системы -10.
Проведём "утряску" воды:
grompp -f pr -c dna_si -p dna -o dna_pr -maxwarn 1
mdrun -deffnm dna_pr -v
Запускаем тестовое моделирование на суперкомпьтере.
ssh skif
cd Djumagulov
grompp -f md -c dna_pr -p dna -o dna_md -maxwarn 1
mpirun -np 16 -q test -maxtime 5 /home/golovin/progs/bin/mdrun_mpi -deffnm dna_md -v
Запускаем основное моделирование на суперкомпьтере:
mpirun -np 16 -maxtime 1200 /home/golovin/progs/bin/mdrun_mpi -deffnm dna_md -v
АНАЛИЗ РЕЗУЛЬТАТОВ
Анализ начинаем с визуального анализа движений молекул:
trjconv -f dna_md.xtc -s dna_md.tpr -o dna_pbc_1.pdb -skip 20 -pbc mol
Файл: dna_pbc_1.pdb
Была получена так же анимация с помощью команды
trjconv -f dna_md.xtc -s dna_md.tpr -o dna_fit_1.pdb -skip 20 -fit rot+trans
в которой изменения ДНК видны лучше. Файл: dna_fit_1.pdb
На мой взгляд образование B-формы лучше происходит в 44 модели, по времени
t= 8800.00000
Однако в следующих фреймах она опять портится. Возможно это из-за того, что у нас относительно небольшой фрагмент ДНК и количество связей не достаточно, чтобы структура сохранялась в одной форма - она "дышит".
Средне-квадратичное отколнение в ходе моделирования
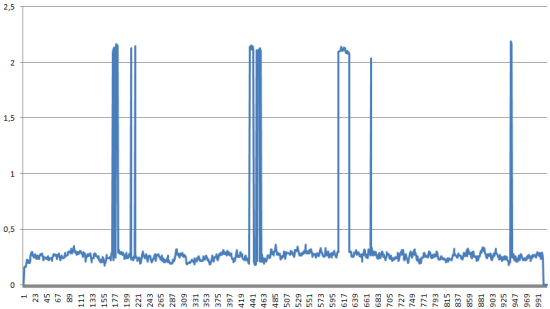
За исключением некоторых точек, сердне-квадратичное отклонение в ходе симуляции достаточно небольшое.
Средне-квадратичное отколнение относительно каждой предыдущей структуры на растоянии 400 кадров. Если ближе к концу закончился конформационный переход, то отколнение должно уменьшаться.
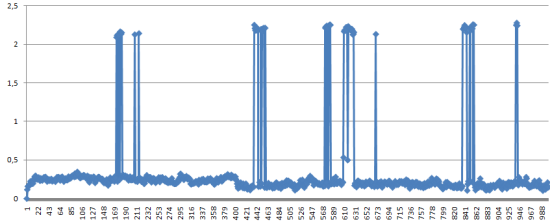
Как и было сказано из наблюдений анимации, структура находится все время в конформационном переходе, если только эти пики не является артифактом моделирования.
Определение изменений гидрофобной и гидрофильной поверхности в ходе конформационного перехода
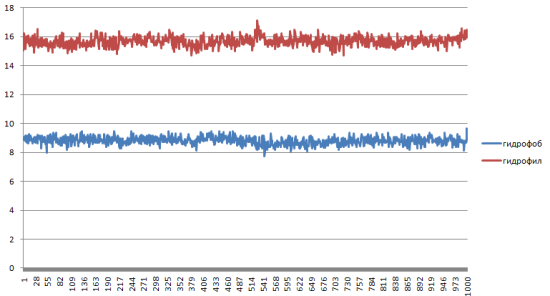
Сильных изменений в гидрофобности и гидрофильности нет.
расчёт колчества образуемых водородных связей. Если мы будем исследовать связи между ДНК и ДНК, то это будут водородные связи между цепями ДНК. Для конца траектории:
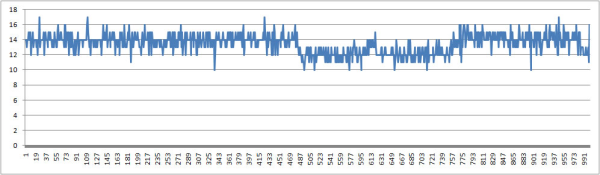
Количество водородных связей все время изменяется. Однако находится в пределах допустимых значений. Что скорее всего связано с неустойчивостью первых и последних п.н. В данной структуре ДНК должно быть 14 водородных связей между цепями.
Количество вдородных связей ДНК-Вода
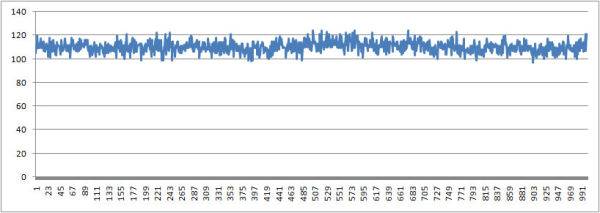
Количество водородных связей ДНК-Вода меняется не сильно. С одной стороны это может быть из-за того, что у нас достаточно не большой фрагмент ДНК, а с другой, что конформационные изменения не сильно влияют на эти связи.
