Построение дуплекса ДНК GATCTA: dna.pdb.
Редактируем полученный от fiber файл: добавим к названиям всех нуклеотидов "D" и заменим "С5М" на "С7".
Построим файл топологии в силовом поле amber99sb и файл с координатами в формате Gromacs:
pdb2gmx -f dna.pdb -o dna -p dna -ff amber99sb -water tip3p
Сделаем небольшой отступ в молекуле ДНК:
editconf -f dna.gro -o dna_ec -d 1.5
Проведём оптимизацию геометрии системы, что бы удалить "плохие" контакты в молекуле:
grompp -f em -c dna_ec -p dna -o dna_em -maxwarn 1
mdrun -deffnm dna_em -v
Step 0. Fmax=4.789e+03
Step 54. Fmax=9.566e+02
Добавим в ячейку молекулу воды:
grompp -f em -c dna_ec -p dna -o dna_em -maxwarn 1
Нейтрализуем заряд :
grompp -f em -p dna -c dna_s -o dna_s
genion -s dna_s -o dna_si -p dna -np 10
Проведём утряску воды:
grompp -f pr -c dna_si -p dna -o dna_pr -maxwarn 1
mdrun -deffnm dna_pr -v
Проанализируем pdb полученные из dna_pr.gro и dna_si.gro:
| dna_pr |
dna_si |
 |
 |
Видно, что расположение молекул в dna_si более упорядоченное, dna_pr их расположение более хаотично. Т.е. после
утряски воды в ячейке, молекулы располагаются хаотично, но при этом ДНК остаётся на месте.
Отправляем файлы на суперкомпьютер (скиф) и, после тестового запуска, запускаем основное моделирование:
mpirun -np 16 -maxtime 1200 /home/golovin/progs/bin/mdrun_mpi -deffnm dna_md -v
Силовое поле используемое при построении топологии.
amber99sb
Заряд системы. Причины этого значения.
0; т.к. мы нейтрализовали заряд -10 после добавления воды, который был на отрицательнозаряженной ДНК. Такой заряд ДНК, скорее всего, обеспечивает количество пар в нашей спирали.
Размер и форму ячейки.
Кубическая ячейка с параметрами: 5.11800 х 4.94600 х 5.49400
Минимизация энергии (из файла mdout.mdp для em.mdp):
o Алогритм минимизации энергии
integrator = l-bfgs ;алгоритм использования малого количества памяти Broyden-Fletcher-;Goldfarb-Shanno (квази-Ньютоновский), метод аппроксимирует Гауссиановскую матрицу из ;пердыдущих конфигураций.
o Алгоритм расчёта электростатики и Ван-дер-Ваальсовых взаимодействий.
Алгоритм для расчёта электростатики - двойное обрезание (cut-off).
Алгоритм объединяет области отсечения со списком neighborlist cut-off (rlist) и отсечения для Кулоновских взаимодействий (Coulomb cut-off) rcoulomb, где rcoulomb ? rlist.
Ван-дер-Ваальсовых взаимодействий - двойное обрезание (cut-off).
Алгоритм объединяет области отсечения со списком neighborlist cut-off (rlist) и отсечения Ван-дер-Ваальсовых взаимодействий (VdW cut-off) rvdw, где rvdw ? rlist.
Модель, которой описывался растворитель
implicit_solvent = No ;т.е. растворителя нет
Утряска растворителя (из файла mdout.mdp для pe.mdp):
o Для биополимеров, укажите параметр который обуславливает неподвижность биополимера.
В строке: define = -DPOSRES. На сколько можно понять, эта строка фиксирует все связи отрицательнозаряженных молекул (через файл posre.itp).
o Число шагов.
nsteps = 10000
o Длина шага.
dt = 0.001 ; в pс
o Алгоритм расчёта электростатики и Ван-дер-Ваальсовых взаимодействий.
Алгоритм для расчёта электростатики - pme (суммирование по Эвальду).
Алгоритм для вычисления VdW - cut-off.
o Алгоритмы термостата и баростата.
Для контроля температуры использовался метод Berendsen. Контроля давления не проиходило.
Основной расчёт МД (из файла mdout.mdp для md.mdp) :
o Время моделирования, количество процессоров, эффективность маштабирования.
4 hours 41 minutes 18 seconds; 16 процессов; 100% эффективность маштабирования
o Длину траектории (=число шагов*длину шага) = 10000 пс.
o Число шагов = 5000000.
o Длина шага = 0,002 пс.
o Алгоритм интегратора - мд.
o Алгоритм расчёта электростатики и Ван-дер-Ваальсовых взаимодействий.
Алгоритм для расчёта электростатики - pme (суммирование по Эвальду). Ван-дер-Ваальсовых взаимодействий - двойное обрезание (cut-off).
o Алгоритмы термостата и баростата.
Для контроля температуры и давления использовался метод Berendsen.
Визуальный анализ движения молекул:
было получено два файла для визауального анализа: dna_pbc_1.pdb и dna_fit_1.pdb.
Более удобный для анализа первый, т.к. две цепи не прыгают по углам ячейки (т.е. ячейка центрирована на положение молекулы).
Молекула изменяет постоянно свою конформацию и на 14 кадре (2800 фсек) переходит в B-форму. А на 30 кадре (6000 фсек) происходит разрыв Т-А пары на 3'-конце.
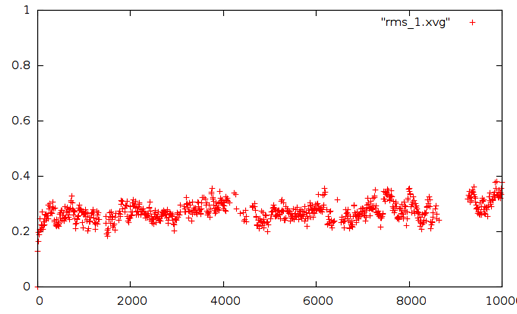
Определение средне-квадратичного отколнения:
| Отклонение от стартовой конформации |
Отклонение относительно каждой предыдущей структуры на расстоянии 400 кадров |
 |
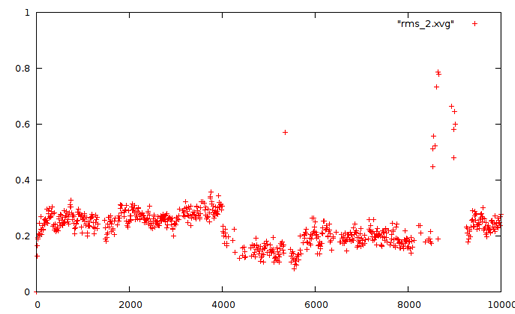 |
Видно, что отклонения от стартовой конформации происходит не сильное, зато относительно предыдущих структур колебание сильнее. Есть даже пик в районе 9000 - там происходит сильное извивание молекулы и в dna_fit_1.pdb видно, что происходит сильный разворот цепей.
Определение изменения гидрофобной и гидрофильной поверхности в ходе конформационного перехода
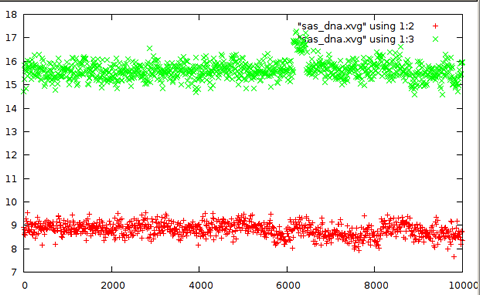
+ - гидрофобная; х - гидрофильная.
Виден скачок гидрофильной поверхности, связанный с разравом связей пары А-Т.
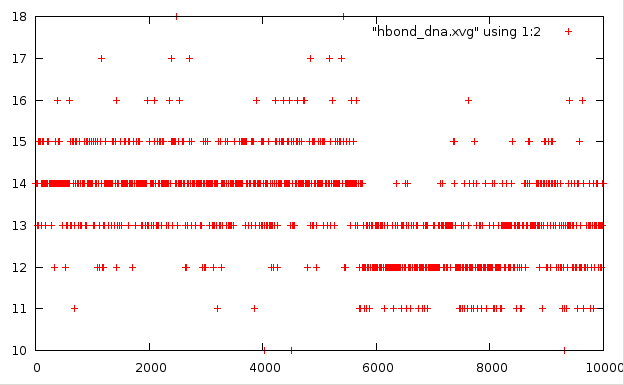
Расчёт колчества образуемых водородных связей ДНК-ДНК и ДНК-вода
| ДНК-ДНК |
ДНК-вода |
 |
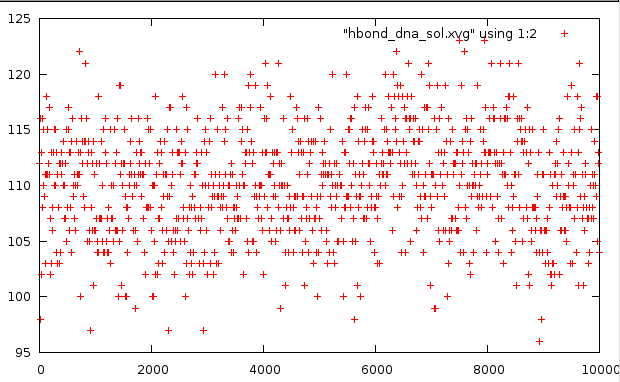 |
Количество водородных связей в дуплексе колеблется от 10 до 18. Канонических связей должно быть 14. Т.е. разброт на плюс-минус 4 связи. При этом в основном всё-таки поддерживается 14. Видно, что на 30 кадре (6000 фсек) произошёл разрыв двух связей (пара А-Т).