Задание 2 (выполнено Борисовой Мариной)
Задача: познакомиться с возможностями Mopac.
I. Cтруктуры молекулы после обработки obgen и оптимизация её с помощью Mopac.
На основе описания порфирина (c1cc2cc3ccc(cc4ccc(cc5ccc(cc1n2)[nH]5)n4)[nH]3) c помощью программы из babel можно построила 3D структуру:
obgen 1.smi > 1.mol
С помощью PyMol удалила ненужные водороды и сохранила результат с расширением pdb. С помощью babel переформатировала координаты в mol формате во входной файл для Mopac.
babel -ipdb 1.pdb -omop 1_opt.mop -xk "PM6"
MOPAC2009.exe 1_opt.mop
Для сравнение переформатировала результат 1_opt.out в pdb:
babel -imopout 1_opt.out -opdb 1_opt.pdb
Провела оптимизацию с параметризацией AM1:
babel -ipdb 1.pdb -omop 1_opt2.mop -xk "AM1"
Для сравнение переформатировала результат 1_opt2.out в pdb:
babel -imopout 1_opt2.out -opdb 1_opt2.pdb
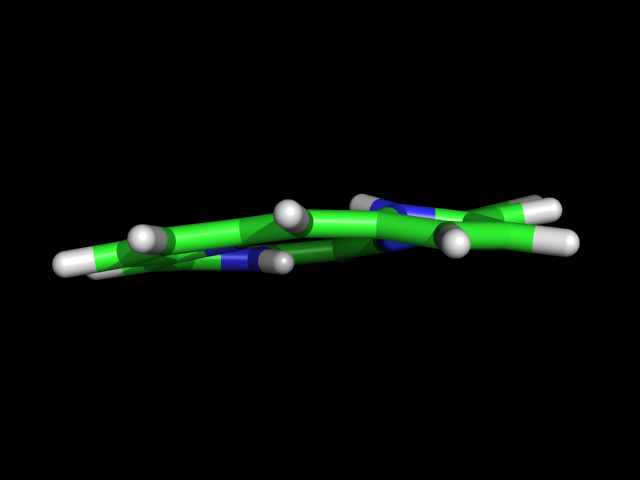
Сравнение структур полученные obgen и Mopac (выводы, картинки и наблюдения):
| obgen |
Mopac (PM6) |
Mopac (AM1) |
 |
 |
 |
| Структура не плоская |
Структура выглядит плоской, но есть небольшой угол между соседними кольцами |
Структура полностью плоская |
Записи "1_opt.mop" и "1_opt2.mop" отличаются DIAGONAL MATRIX и энергиями. При оптимизации с параметром "PM6" энергия (FINAL HEAT OF FORMATION) меньше, а также есть различия в других энаргиях. Оптимизация с параметром "AM1" происходила несколько дольше.
Получается, что с параметром "AM1" молекула выглядит более правильной, т.е. оптимизация проходит лучше.
II. Рассчёт возбуждённых состояний порфирина и спектр его поглощения.
Для указания Mopac о необходимости расчёта возбуждённого состояния добавила в конец файла:
пустую строку
cis c.i.=4 meci oldgeo
some description
MOPAC2009.exe 1_opt_spect.mop
В конце файла нашла значения энергий для электронных переходов. И рассчитала длину волн при которых происходят эти переходы (wave length = h*c/ENERGY):
| STATE |
ENERGY |
SPIN |
wave length |
| 1+ |
0 |
SINGLET |
|
| 2 |
1,91 |
TRIPLET |
647,66 |
| 3 |
2,27 |
SINGLET |
546,97 |
| 4 |
2,46 |
TRIPLET |
503,16 |
| 5 |
2,83 |
TRIPLET |
438,78 |
| 6 |
3,36 |
TRIPLET |
368,53 |
| 7 |
3,39 |
SINGLET |
365,58 |
| 8 |
3,67 |
SINGLET |
337,91 |
| 9 |
3,87 |
SINGLET |
320,21 |
III. Определение геометрии молекулы и её дианиона O=C1C=CC(=O)C=C1 с помощью obgen и Мopac.
Определила геометрию молекулы с помощью obgen и Мopac (оптимизация проводилась с параметром "AM1", т.к. в предыдущем задании этот вариант показал результаты более близкие к реальности):
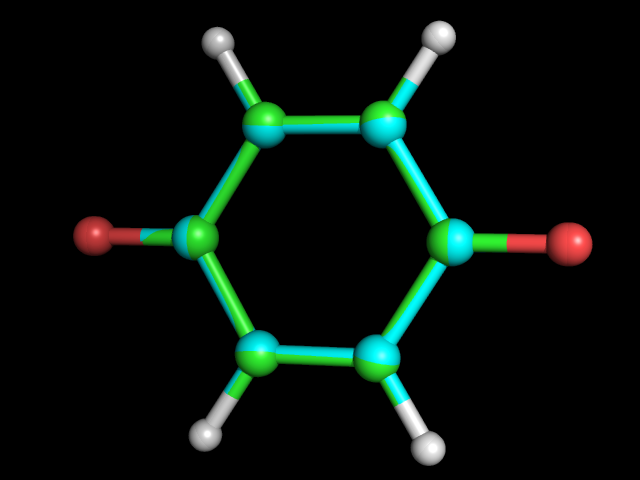
Структуры полученные с помощью: зелёные углероды - obgen, голубые - Mopac. |
Видно, что атомы углерода, которые связаны с кислородами, в структуре obgen находятся ближе к центру цикла. Т.е. структура Mopac получается немного вытянутой к кислородам и сплюснута с остальных боков. |
Определила геометрию дианиона молекулы (предварительно в mop файл была вставлена запись CHARGE=-2 и внесены изменения в атомы, имеющие отрицательный заряд):
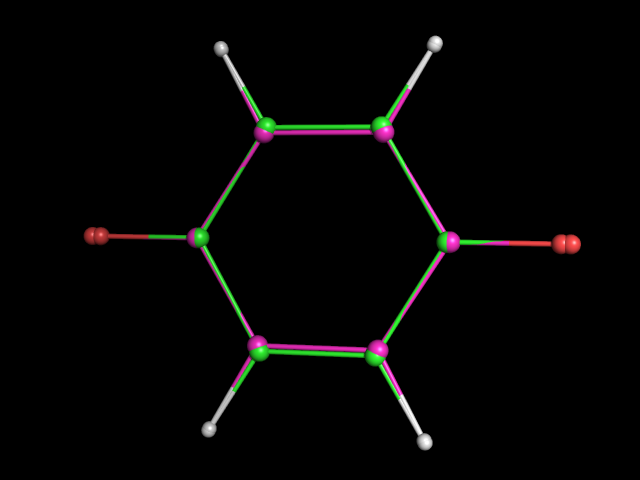
Структуры полученные с помощью: зелёные углероды - obgen, малиновые - Mopac (дианион). |
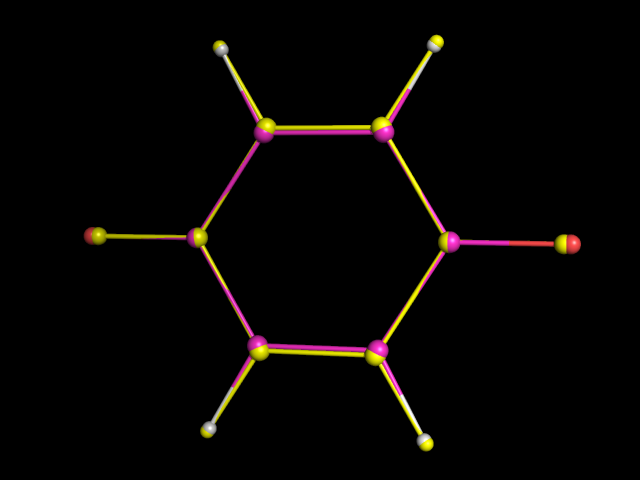
Струтуры полученые с помощью Mopac: жёлтые атомы - нейтральная молекула, малиновые углероды - дианион. |
Видно, что струтура дианиона ещё больше вытянута в стороны кислородов. Скорее всего это происходит в связи с тем, что кислороды дианиона больше не связывают двойные связи с углеродами - они отодвигаются дальше от кольца. Цикл становится более ароматическим.
babel -ipdb test.pdb -opdb test1.pdb -p 7,4
- добавление магния: прописываем гетероатом магния (62-ой в файле) и присваиваем координаты (среднее арифметическое между координатами атомов PG и CA)
- "замораживание атомов": меняем у всех атомов, кроме гамма-фосфата, воды и магния, значения около координат ("1"->"0")
- оптимизация структуры (test2.pdb)
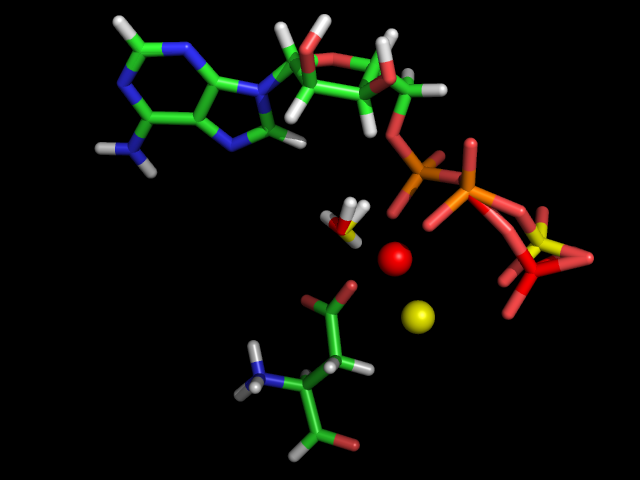
Оптимизированные атомы отмечены жёлтым цветом + атомы связанные с ними напрямую. Неоптимизированные атомы - красные. |
После оптимизации магний расположился ближе к кислородам, причём гамма-фосфат передвинулся так, чтобы его кислород находился на равном с другими фосфатными кислородами расстоянии от магния. Атомы воды развернулись кислородом к магнию. Поэтому естественно предположить, что пять кислородов координируют атом магния (три фосфатных и аспартатный, кислородом воды).
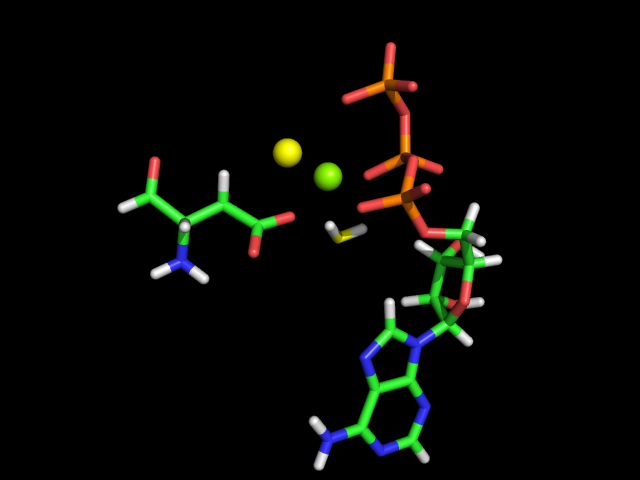
Неоптимизированная (жёлтые атомы) и 3pp1 структуры. |
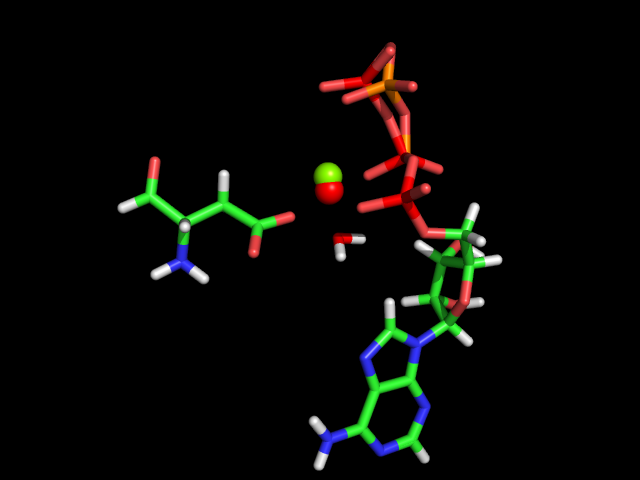
Оптимизированная (красные атомы) и 3pp1 струтура. |
Видно, в 3pp1 структура отличается от обоих структур (неоптимизированной и оптимизированной). Фосфатные атомы располагаются как в неоптимизированной структуре, но атом магния располагается, как в оптимизированной. Что кажется странным, т.к. возникает вопрос в каком положении фосфаты на самом деле координируют атом магния. Если верна 3рр1 структура, значит оптимизация не дала правильного представления о положении соединения.
© 2010-2011 Borisova Marina