Занятие 4.
Вычисление параметров для молекулярной механики.
Суть задания состоит в определении констант ковалентных взаимодействий для молекулярной механики на основе квантово-химических расчётов.
-
У нас есть оптимизированная структура этана в виде z-matrix:
$DATA eth C1 C C 1 cc H 2 ch 1 cchv H 2 ch 1 cch 3 d1 0 H 2 ch 1 cch 3 d2 0 H 1 ch 2 cch 3 d3 0 H 1 ch 2 cch 5 d3 0 H 1 chv 2 cch 4 d3 0 cc=1.52986 ch=1.08439 chv=1.08439 cch=111.200 cchv=111.200 d1=120 d2=-120 d3=180 $END
Вместо значений длин и углов связей стоят переменные. Необходимо создать порядка 20 разных файлов для расчёта энергии в Gamess с разными значениями по длине одной из связей. Для этого в представленных координатах одна связь или угол отличается названием переменной, но не её значением. Автоматизируем процесс с помощью скрипта на bash.
-
Сначала создадим файл-заготовку для размножения et.inp.
Для этого к координатам добавим шапку для DFT из предыдущего практикума.
Но нам нужно изменить информацию о типе входных координат: заменим COORD=CART на COORD=ZMT.
Для того, чтобы проверить, работает ли файл-заготовка, запустим GAMESS => получаем файл et.log без ошибок. Идем дальше!
-
Теперь создадим текстовый файл скрипта make_b.bash со следующим содержанием:
#!/bin/bash ### делаем цикл от -10 до 10 ##### for i in {-10..10}; do #### нам надо рассчитать новую длину связи ##### #### с шагом 0.02 ангстрема, ##### #### воспользуемся калькулятором bc ##### #### и результат поместим в переменную nb ##### nb=$(echo "scale=5; 1.52986 + $i/50" | bc -l) #### пролистаем файл et.inp и заменим указание переменной ### #### на новое значение и пере направим результат в файл ### sed "s/cc=1.52986/cc=$nb/" et.inp > b_${i}.inp doneНужно было поправить скрипт так, чтобы стартовая длина изменяемой связи соответствовала файлу et.inp). 1.52986 - стартовая длина изменяемой связи.
Запускаем скрипт :bash ./make_b.bash
В итоге я получила 21 inp файл и в каждом - разное значение для переменной сс.
-
Теперь запустим расчёт для этих файлов.
Для этого перед строчкойdone
вставим запуск Gamess:gms b_${i}.inp 1 > b_${i}.logЗапускаем скрипт.
Теперь необходимо извлечь значение энергии из log файла. Удобно воспользоваться awk.
Сначала в скрипте прокомментируем запуск Gamess, поставив в начало строчки c gms #.gms b_${i}.inp 1 > b_${i}.log####.
Добавим после этой строчки вызов awk, при этом мы ищем строчку с TOTAL ENERGY и печатаем четвертое поле, считая, что поля разделены пробелами:awk '/TOTAL ENERGY =/{print $4}' b_${i}.logЗапускаем скрипт. На экране появляется 21 значение энергии. Теперь удобно было бы выводить и значение длины связи. Для этого добавим перед вызовом awk распечатку переменой nb. Распечатаем переменную и несколько пробелов без переноса строки:echo -n "$nb "
Далее перенаправим поток вывода скрипта в файл bond:bash ./make_b.bash > bond
В результате имеем файл bond с двумя столбцами (с длинами связи и соответствующими значениями энергии).
-
У нас есть зависимость энергии молекулы от длины одной связи. Построим эту зависимость в gnuplot.
Для этого запустим Xming->XLaunch. Выберем тип расположения окон, удобно использовать Multiple windows. Next. Выберем Start Program. Run Remote-> Putty.
Дальше всё как обычно: kodomo, username.
Перейдем в рабочую директорию. Запустим Gnuplot:gnuplot
Построим зависимость энергии от длины связи, просто введем : plot "bond"
В итоге получаем график с точками, похожими на параболу. Теперь надо найти коэффициенты в функции f(x)=a+k(x-b)^2 которые бы позволили наиболее близко описать наблюдаемую зависимость. Для этого воспользуемся возможностями Gnuplot. Сначала зададим функцию в развернутом виде, в строке gnuplot введём:f(x)=a + k*x*x - 2*k*x*b + k*b*b
И зададим стартовые значения коэффициентов:a=-80 k=1 b=1.5
Проведём подгонку коэффициентов под имеющиеся точки в файле bond:fit f(x) "bond" via a,k,b
Сохраним значения коэффициентов в файле koeff.
Построим графики функции и значений энергии из Gamess:plot "bond", f(x)
Изображение полученного графика: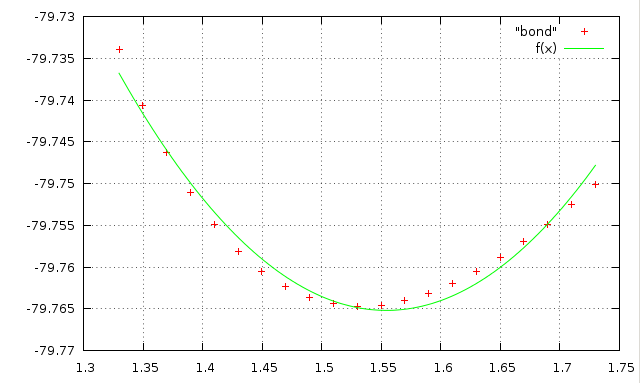
Причиной неточного совпадения точек и функции возможно является большая ошибка в определении параметра k.
-
Проделаем аналогичные операции для валентного угла HCH (cch). Будем изменять его значения от 109.2 до 113.2 с шагом 0.2. В результате получим файл со значениями угла и соответствующими им энергиями.
Параметры подгонки.
График зависимости энергии от угла:
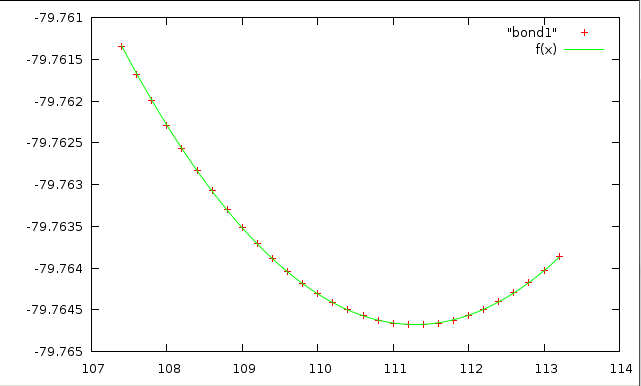
Полученная апроксимация хорошая - все точки лежат прямо на кривой.
-
Проделаем аналогичные операции для торсионного угла d3. Будем изменять его значения от -180 до 180 с шагом 12. В результате получим файл со значениями угла и соответствующими им энергиями.
График зависимости энергии от угла:
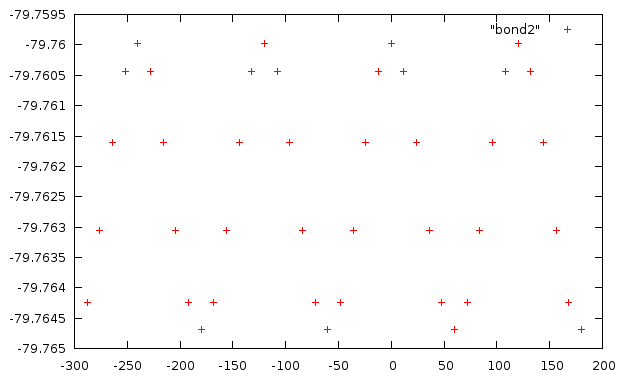
Подгонку не проводили. Эта функция имеет 3 минимума.
<<Обратно на шестой семестр
<<Обратно на главную страницу
©Лелекова Мария,2011