Занятие 2
Cеми-эмпирические вычисления: Mopac.
Занятие заключается в поэтапном освоении возможностей Mopac как пакета для оптимизации структуры молекул и расчёта некоторых свойств.
Информацию о Mopac можно прочитать здесь. Помощь к программе babel с помощью putty: babel -h.
Сначала необходимо установить переменные:
export PATH=${PATH}:/home/preps/golovin/progs/bin
export MOPAC_LICENSE=/home/preps/golovin/progs/bin
Задание 1.
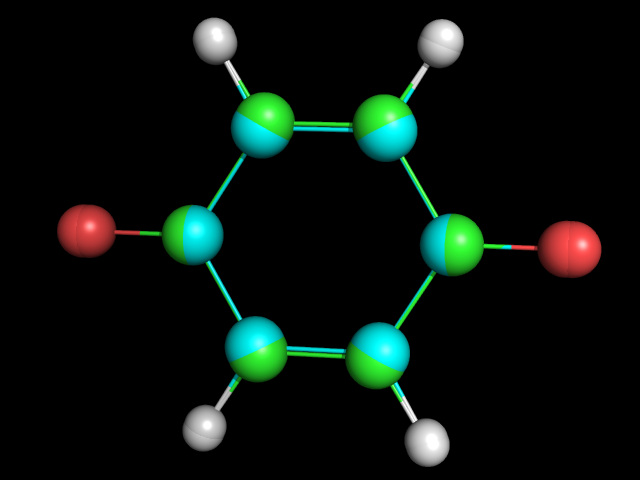
Я узнала аннотацию порфирина в виде SMILES на этом сайте. Создала файл 1.smi, в нем сначала идут строки SMILES порфирина, а через несколько пробелов просто porphyrin.
На основе этого описания c помощью программы из babel построила 3D структуру порфирина:
obgen 1.smi > 1.molЗатем я рассмотрела полученную структуру в PyMol:
Полученное изображение:
С помощью babel переформатировала координаты в mol формате во входной файл для Mopac:
babel -ipdb 1.pdb -omop 1_opt.mop -xk "PM6"
С помощью -xk мы задала параметризацию типа pm6.
Далее запустила Mopac для файла:
MOPAC2009.exe 1_opt.mop
Затем изучила файл вывода out. Для сравнения переформатировала результат в pdb:
babel -imopout 1_opt.out -opdb 1_opt.pdb
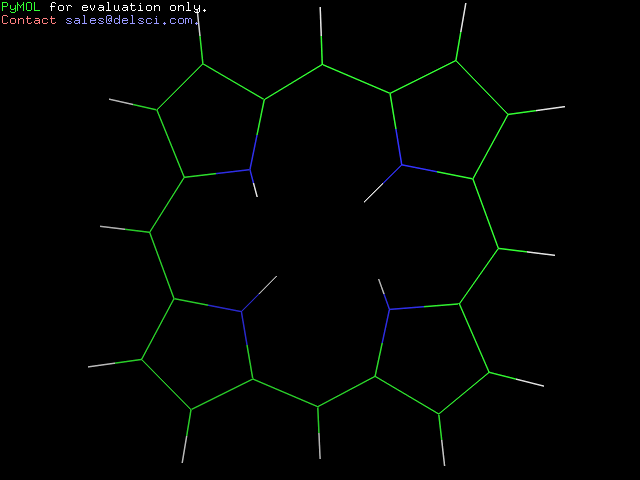
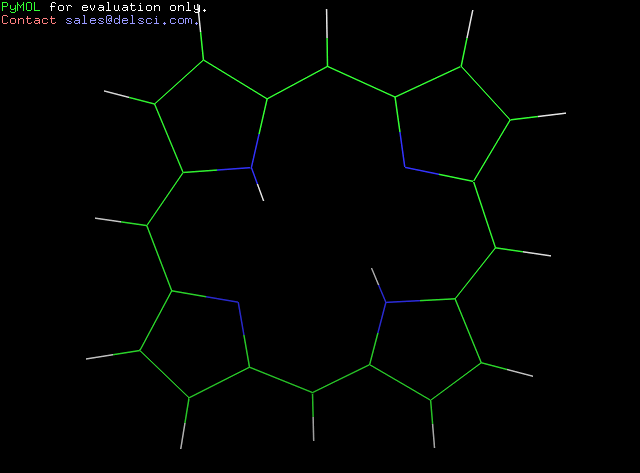
Получила следующее изображение:
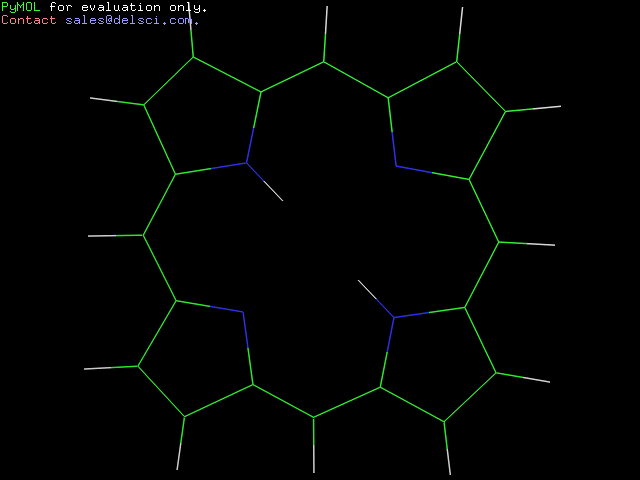
Можно увидеть, что с помощью obgen и Mopac получились разные структуры:
|
|
|
Вывод на основе наблюдения: Mopac строит структуру такой, какой она является на самом деле.
Задание 2.
Мне нужно было рассчитать возбужденные состояния порфирина и на основе этих данных оценить спектр поглощения молекулы.
Для расчёта возбуждённых состояний я использовала файл из предыдущего задания.
Для того, чтобы указать Mopac о необходимости расчёта возбуждённого состояния, необходимо добавить в конец файла:
пустую строку cis c.i.=4 meci oldgeo some descriptionДля запуска Mopac ввела команду:
MOPAC2009.exe 1_opt_spectr.mop
В конце файла находятся значения энергий для электронных переходов:
STATE ENERGY (EV) Q.N. SPIN SYMMETRY POLARIZATION
ABSOLUTE RELATIVE X Y Z
1+ 0.000000 0.000000 1+ SINGLET ????
2 1.913312 1.913312 1 TRIPLET ????
3 2.266014 2.266014 2 SINGLET ????
4 2.463186 2.463186 2 TRIPLET ????
5 2.823915 2.823915 3 TRIPLET ????
6 3.362161 3.362161 4 TRIPLET ????
7 3.389757 3.389757 3 SINGLET ???? 0.2031 0.2347 0.0010
8 3.669242 3.669242 4 SINGLET ???? 2.3899 2.0438 0.0085
9 3.871323 3.871323 5 SINGLET ???? 1.5461 1.7992 0.0084
Можно рассчитать длины волн, при которых происходят эти переходы ( Е=h*c/длина волны ):Энергия (эВ) Длина волны (нм) 1,913312 648,913002 2,266014 547,910575 2,463186 504,0516769 2,823915 439,6637412 3,362161 369,2782808 3,389757 366,2719876 3,669242 338,3731664 3,871323 320,7102672Задание 3.
Для молекулы O=C1C=CC(=O)C=C1 необходимо было определить геометрию как с помощью obgen, так и Мopac.
Нужно было определить геометрию дианиона этой молекулы.Я работала с mop-файлом для Mopac, получила файл-pdb.
Полученные структуры, наложенные друг на друга (зеленым цветом представлены атомы углерода из файла
для Мopac, а голубым - для obgen):
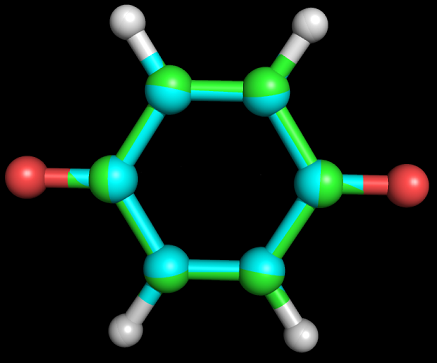
Видно, что молекулы идентичны.
В первую строчку mop файла добавила слово CHARGE=-2.
Потом указала на каких атомов должен находиться отрицательный заряд.Полученный файл.
Проведем сравнение полученнх структур, наложенных друг на друга (атомы углерода для первой структуры зеленые, а для второй - голубые):