Cеми-эмпирические вычисления: Mopac
Работу нужно начинать с установки переменных:
export PATH=${PATH}:/home/preps/golovin/progs/bin
export MOPAC_LICENSE=/home/preps/golovin/progs/bin
-
Найдем в интернете порфирин и его аннотацию в виде SMILES
(например здесь ).
� Создадим текстовый файл 1.smi где сначала строки SMILES порфирина
а через несколько пробелов просто porphyrin. На основе такого опсания можно построить
3D структуру порфирина с помощью программы babel:
obgen 1.smi > 1.mol
� Удалим ненужные водороды и посмотрим результат в PyMol:
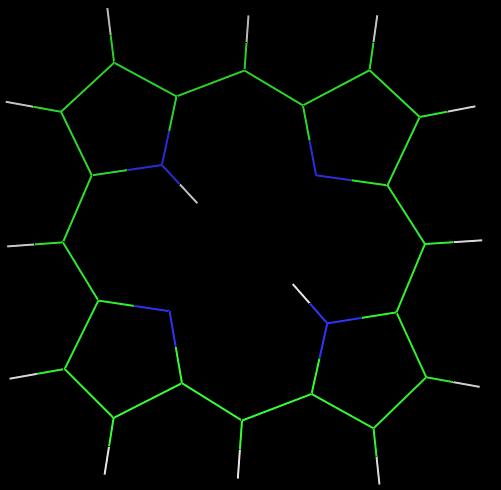
Теперь переформатируем координаты в mol формате во входной файл для Mopac:
babel -ipdb myfile.pdb -omop 1_opt.mop -xk "PM6"
*c помощью -xk мы задали параметризацию типа pm6
Запустим Mopac :
MOPAC2009.exe 1_opt.mop
Теперь сравним результаты obgen и mopac. Для этого переформатируем
выходной файл Mopac в pdb:
babel -imopout 1_opt.out -opdb 1_opt.pdb
Сравним:


Как мы видим, MOPAC, в отличие от babel, построил плоскую структуру.
� - Рассчитаем возбужденные состояния порфирина и на основе этих данных
прикинем спектр поглощения молекулы.
Для расчёта возбуждённых состояний копируем mop файл из предыдущего занятия.
Для указания Mopac о необходимости расчёта возбуждённого состояния добавим в конец файла:
пустую строку и строки:
cis c.i.=4 meci oldgeo
some description
Запустим Mopac:
MOPAC2009.exe 1_opt_spectr.mop
� На выходе получаем файл 1_opt_spectr.out.
В конце файла значения энергий для электронных переходов:
STATE ENERGY (EV) Q.N. SPIN SYMMETRY POLARIZATION
ABSOLUTE RELATIVE X Y Z
1+ 0.000000 0.000000 1+ SINGLET ????
2 1.913312 1.913312 1 TRIPLET ????
3 2.266014 2.266014 2 SINGLET ????
4 2.463186 2.463186 2 TRIPLET ????
5 2.823915 2.823915 3 TRIPLET ????
6 3.362161 3.362161 4 TRIPLET ????
7 3.389757 3.389757 3 SINGLET ???? 0.2031 0.2347 0.0010
8 3.669242 3.669242 4 SINGLET ???? 2.3899 2.0438 0.0085
9 3.871323 3.871323 5 SINGLET ???? 1.5461 1.7992 0.0084
The "+" symbol indicates the root used.
� На основании этих значений и простой формулы E = h*v рассчитаем длину волн при которых
происходят эти переходы:
Энергия (эВ)
Длина волны (нм)
1,913312
648
2,266014
546
2,463186
504
2,823915
439
3,362161
369
3,389757
365
3,669242
338
3,871323
320
- Для молекулы O=C1C=CC(=O)C=C1 определите геометрию как с помощью obgen так и Мopac
(см. выше). Получаем:
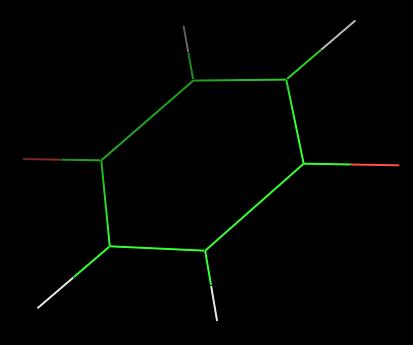
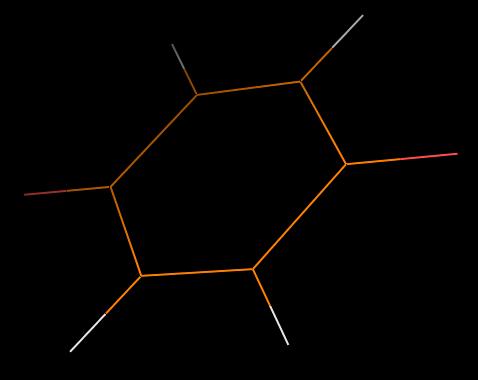
Как мы видим, молекулы получились абсолютно идентичные.
Теперь определим геометрию дианиона этой молекулы.
Для этого в первую строчку mop файла добавляем слово CHARGE=-2.
Потом явным способом указываем на каких атомах должен находиться отрицательный заряд. Пример:
PM6 CHARGE=-2 gg O(-) 0.98570 1 0.00130 1 -0.43680 1 C 2.16830 1 0.00680 1 -0.12400 1 ...........
Рассмотрим наложение получившихся структур:
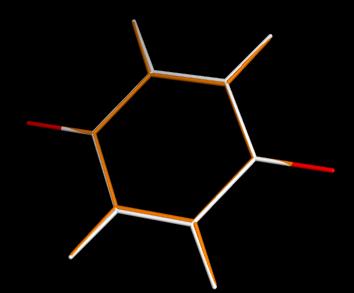
*белым - незаряженная молекула; оранжевым - заряженная
Структуры отличаются по расположению связей. Заряженная структура незначительно
сжата к оси кислородов, связи C-O вытянулись у заряженной молекулы из-за появления ароматичности.
© Азнаурян 2008
marina-91@list.ru
export PATH=${PATH}:/home/preps/golovin/progs/bin
export MOPAC_LICENSE=/home/preps/golovin/progs/bin obgen 1.smi > 1.mol
� Удалим ненужные водороды и посмотрим результат в PyMol:
Теперь переформатируем координаты в mol формате во входной файл для Mopac:
babel -ipdb myfile.pdb -omop 1_opt.mop -xk "PM6"
*c помощью -xk мы задали параметризацию типа pm6
Запустим Mopac :
MOPAC2009.exe 1_opt.mop
Теперь сравним результаты obgen и mopac. Для этого переформатируем выходной файл Mopac в pdb:
babel -imopout 1_opt.out -opdb 1_opt.pdb
Сравним:
|
|
|
Как мы видим, MOPAC, в отличие от babel, построил плоскую структуру.
cis c.i.=4 meci oldgeo
some description
Запустим Mopac:
MOPAC2009.exe 1_opt_spectr.mop
� На выходе получаем файл 1_opt_spectr.out. В конце файла значения энергий для электронных переходов:
STATE ENERGY (EV) Q.N. SPIN SYMMETRY POLARIZATION
ABSOLUTE RELATIVE X Y Z
1+ 0.000000 0.000000 1+ SINGLET ????
2 1.913312 1.913312 1 TRIPLET ????
3 2.266014 2.266014 2 SINGLET ????
4 2.463186 2.463186 2 TRIPLET ????
5 2.823915 2.823915 3 TRIPLET ????
6 3.362161 3.362161 4 TRIPLET ????
7 3.389757 3.389757 3 SINGLET ???? 0.2031 0.2347 0.0010
8 3.669242 3.669242 4 SINGLET ???? 2.3899 2.0438 0.0085
9 3.871323 3.871323 5 SINGLET ???? 1.5461 1.7992 0.0084
The "+" symbol indicates the root used.
� На основании этих значений и простой формулы E = h*v рассчитаем длину волн при которых
происходят эти переходы:
| Энергия (эВ) | Длина волны (нм) |
| 1,913312 | 648 |
| 2,266014 | 546 |
| 2,463186 | 504 |
| 2,823915 | 439 |
| 3,362161 | 369 |
| 3,389757 | 365 |
| 3,669242 | 338 |
| 3,871323 | 320 |
|
|
|
Как мы видим, молекулы получились абсолютно идентичные.
Теперь определим геометрию дианиона этой молекулы. Для этого в первую строчку mop файла добавляем слово CHARGE=-2. Потом явным способом указываем на каких атомах должен находиться отрицательный заряд. Пример:
PM6 CHARGE=-2 gg O(-) 0.98570 1 0.00130 1 -0.43680 1 C 2.16830 1 0.00680 1 -0.12400 1 ...........Рассмотрим наложение получившихся структур:
*белым - незаряженная молекула; оранжевым - заряженная
Структуры отличаются по расположению связей. Заряженная структура незначительно сжата к оси кислородов, связи C-O вытянулись у заряженной молекулы из-за появления ароматичности.
© Азнаурян 2008 marina-91@list.ru