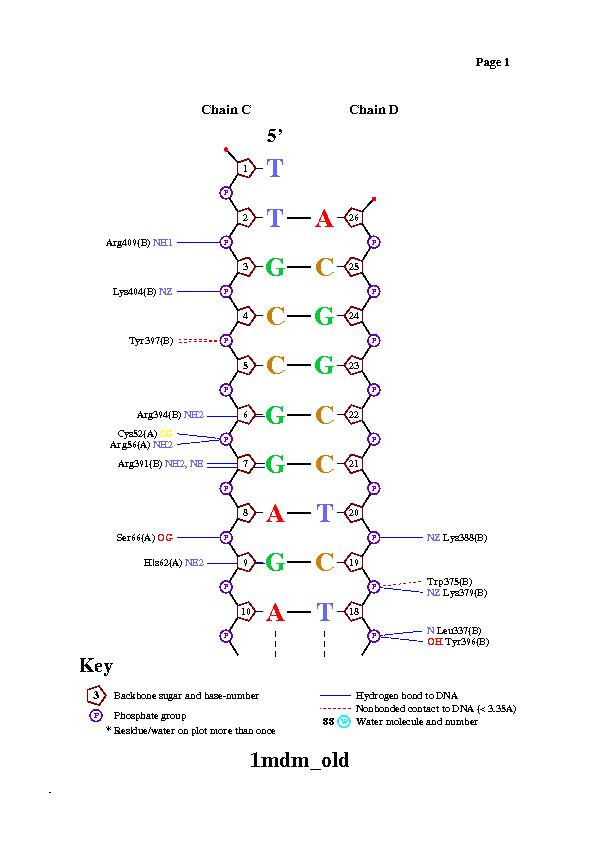
| Контакты атомов белка с | Полярные | Неполярные | Всего |
| остатками 2'-дезоксирибозы | 10 | 4 | 15 |
| остатками фосфорной кислоты | 23 | 0 | 23 |
| остатками азотистых оснований со стороны большой бороздки | 6 | 3 | 9 |
| остатками азотистых оснований со стороны малой бороздки | 5 | 0 | 5 |
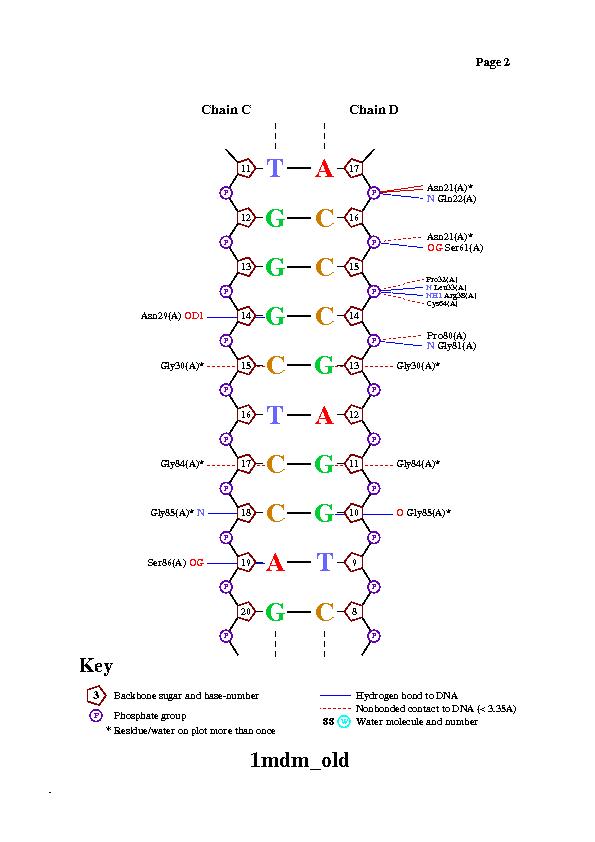
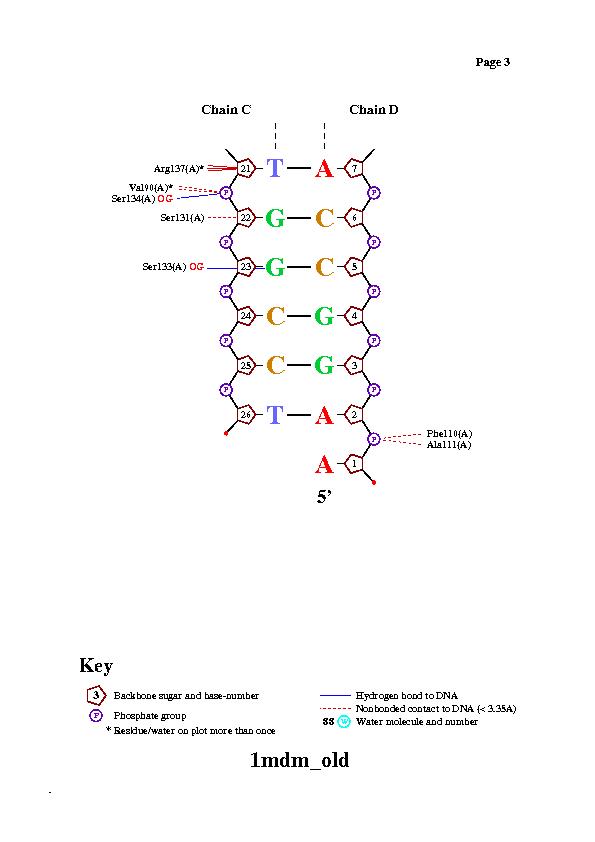
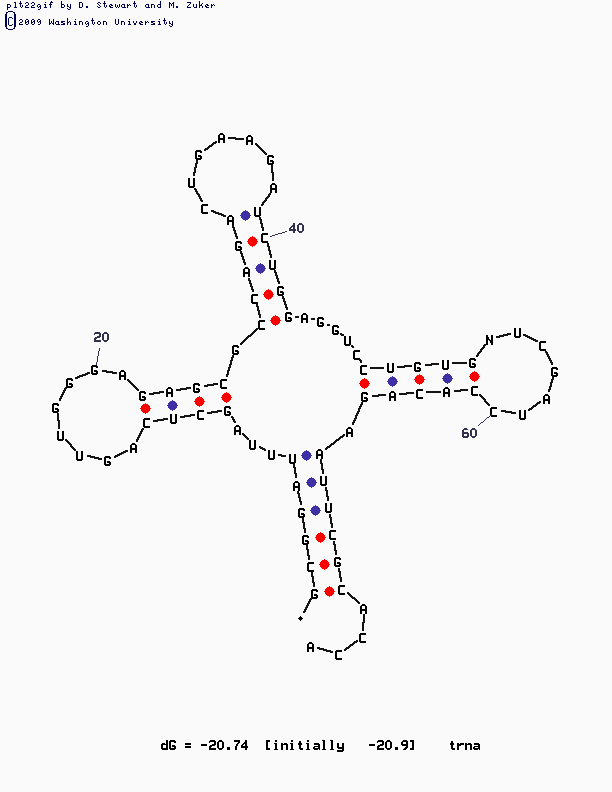
| Участок структуры | Позиции в структуре (по результатам find_pair) |
Результаты предсказания с помощью einverted |
Результаты предсказания по алгоритму Зукера |
| Акцепторный стебель | (5') 1-7 (3') (5') 66-72 (3') Всего 7 пар |
предсказано 0 пар | предсказано 6 пар из 7 (кроме 7-66) |
| D-стебель | (5') 10-13 (3') (5') 22-25 (3') всего 4 пары |
предсказано 0 пар | все 4 пары предсказаны |
| T-стебель | (5') 49-52 (3') (5') 62-65 (3') всего 4 пары |
предсказано 5 пар (дополнительная 53-61) | предсказаны все 4 пары + 1 лишняя (53-61) |
| Антикодоновый стебель | (5') 39-43 (3') (5') 27-31 (3') всего 5 пар |
предсказано 5 пар | все 5 пар предсказаны |
| Общее число канонических пар нуклеотидов | 20 | 10 | 20 |