Структура тРНК
Структура тРНК
1)Краткое описание структуры в файле 1EVV.pdb
На сайте PDB я получил структуру заданной РНК (идентификатор 1evv). Также я нашёл краткое описание структуры.
Молекула получена из организма дрожжей Saccharomyces.
Для исследования была выбрана цепь A, представляющая фенилаланиновую тРНК со следующей последовательностью:
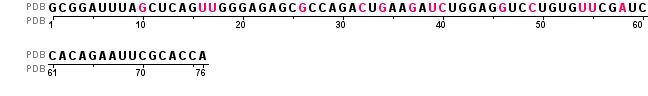
[1] 5' - G C G G A U U U A 2MG C U C
A G H2U H2U G G G A G A G C M2G
C C A G A OMC U OMG A A YG A PSU
5MC U G G A G 7MG U C 5MC U G U
G 5MU PSU C G 1MA U C C A C A G
A A U U C G C A C C A - 3' [76],
где
901 и 970 - номера первого и последнего нуклеотида.
Длина цепи 76 оснований. Красным отмечены модифицированые основания. На 3' конце есть триплет CCA, к которому присоединяется аминокислота изолейцин.
2)Исследование вторичной структуры
С помощью программы find_pair пакета 3DNA были определены возможные водородные связи
между азотистыми основаниями (1evv.out). В соответствии с полученными данными
акцепторный стебель состоит из участка 1-7 и комплементарного ему участка 72-66.
Т-стебель - из участков 49-53 и 65-61
D-стебель - из участков 10-13 и 25-22
антикодоновый стебель - из участков 38-44 и 32-26
На картинке изображена т-РНК в остовной модели.
Акцепторный стебель выделен красным, T-стебель зеленым,
D-стебель синим и антикодоновый стебель оранжевым.
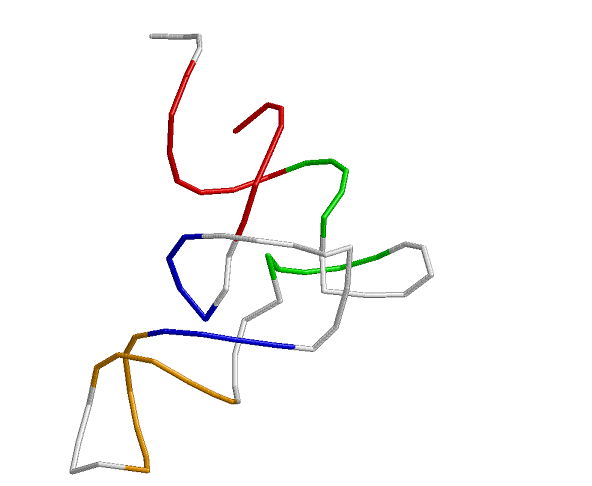
Рис.1. Вторичная структура молекулы фенилаланиновой тРНК из SACCHAROMYCES
|
Скрипт для получения изображения:
background white
restrict none
select all
backbone 100
color white
select 1-7
or 66-72
color red
select 49-53
or 61-65
color green
select 10-13
or 22-25
color blue
select 38-44
or 26-32
color orange
|
Фрагмент файла, полученного командой find_pairs:
Strand I Strand II Helix
1 (0.003) A:...1_:[..G]G-----C[..C]:..72_:A (0.005) |
2 (0.004) A:...2_:[..C]C-----G[..G]:..71_:A (0.006) |
3 (0.008) A:...3_:[..G]G-----C[..C]:..70_:A (0.006) |
4 (0.009) A:...4_:[..G]G-*---U[..U]:..69_:A (0.007) |
5 (0.009) A:...5_:[..A]A-----U[..U]:..68_:A (0.008) |
6 (0.007) A:...6_:[..U]U-----A[..A]:..67_:A (0.016) |
7 (0.007) A:...7_:[..U]Ux---xA[..A]:..66_:A (0.012) x
8 (0.008) A:..41_:[..U]U-----A[..A]:..29_:A (0.006) |
9 (0.012) A:..42_:[..G]G-----C[..C]:..28_:A (0.005) |
10 (0.011) A:..43_:[..G]Gx---xC[..C]:..27_:A (0.006) |
11 (0.014) A:..45_:[..G]Gx*---C[..C]:..25_:A (0.006) |
12 (0.006) A:..11_:[..C]C-----G[..G]:..24_:A (0.017) |
13 (0.009) A:..12_:[..U]U-----A[..A]:..23_:A (0.013) |
14 (0.011) A:..13_:[..C]C----xG[..G]:..22_:A (0.011) |
15 (0.017) A:..14_:[..A]A-**-xU[..U]:...8_:A (0.008) |
16 (0.018) A:..15_:[..G]Gx**+xC[..C]:..48_:A (0.013) x
17 (0.018) A:..19_:[..G]Gx---xC[..C]:..56_:A (0.012) +
18 (0.008) A:..33_:[..U]Ux*--xA[..A]:..36_:A (0.007) +
19 (0.015) A:..50_:[..U]U-----A[..A]:..64_:A (0.015) |
20 (0.011) A:..51_:[..G]G-----C[..C]:..63_:A (0.009) |
21 (0.014) A:..52_:[..U]U-----A[..A]:..62_:A (0.022) |
22 (0.015) A:..53_:[..G]G-----C[..C]:..61_:A (0.006) |
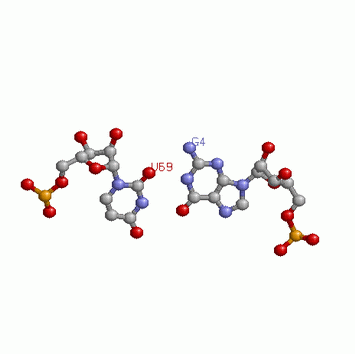
Структуру стеблевых дуплексов поддерживают 21 канонических и 1 неканонической пары оснований..
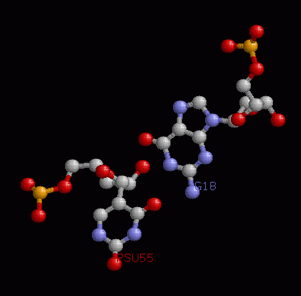
Ниже приведено изображение неканонической пары. Это остатки G4 и U69
а)в структуре нет вариабельной петли;
а)в T-петле отсутствует остаток тимидинна;
а)в D-петле отсутствует остаток дигидроуридина.
3)Исследование третичной структуры
1. Данные о предположительных стрекинг-взаимодействиях получены в результате выполнения команды:
find pair -t 1EVV.pdb stdout | analyze и содержатся в выходном файле - 1EVV.out.
Всего было выдано 28 потенциально возможных взаимодействий.
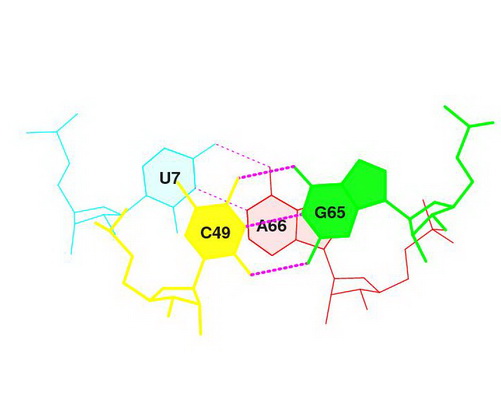
Перекрытие Uc/GA, обладающее площадью (4,19 квадратных ангстрем), было замечено под номером 7.
Это - стрекинг-взаимодействие между основаниями U7 и A66 конца акцепторного стебля и с49 и G65 начала T-стебля.
Изображение соответствующего стекинг-взаимодействия получено при помощи выполнения последовательных команд:
ex_str -7 stacking.pdb step7.pdb (вырезание нужной структуры в отдельный файл),
stack2img -cdolt step7.pdb step7.ps (построение изображения).
2. Среди шести водородных взаимойдейсвий вне стеблей я выделил три, которые происходят между основаниями D и Т-петель.
Это пары нуклеотидов
U54 - a58
U55 - G18
G15 - C48
Неканоническая среди них только одна U55 - G18.
3) Предсказание вторичной структуры тРНК
Программа einverted.
Вводимая нуклеотидная последовательность: 1EVV.fasta
Штраф за гэп [12]: 4
(именно при этом значении получились наиболее адекватные р-ты)
Минимальное значение порога [50]:15
Пробовались значения 20,50 (улучшения эти значения не принесли)
Значение основания (канонической пары) [3]:3
Значение неканонической пары("несоответствие") [-4]:-4
Выходной файл :1EVV.inv
Выравнивание
1EVV: Score 16: 8/10 ( 80%) matches, 0 gaps
22 gagcgccaga 31
|| | |||||
48 ctggaggtct 39
1EVV: Score 15: 5/5 (100%) matches, 0 gaps
49 ctgtg 53
|||||
65 gacac 61
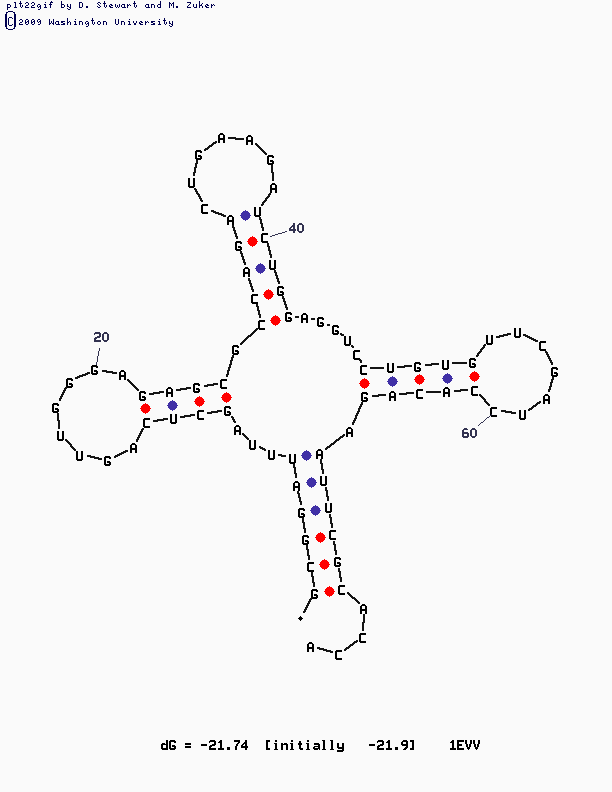
Реальная и предсказанная вторичная структура тРНК из файла 1EVV.pdb
| Участок структуры |
Позиции в структуре (по результатам find_pair) |
Результаты предсказания с помощью einverted |
Результаты предсказания по алгоритму Зукера |
| Акцепторный стебель |
5' 1-7 3'
5' 66-72 3'
Всего 7 пар
|
Предсказано 0 пар из 7 реальных |
Предсказано 6 пар из 7 реальных |
| D-стебель |
5' 10-13 3'
5' 22-25 3'
Всего 4 пары
|
Предсказано 0 пар из 4 реальных |
Предсказано 4 пары из 4 реальных |
| T-стебель |
5' 49-53 3'
5' 61-65 3'
Всего 5 пар
|
Предсказано 5 пар из 5 реальных |
Предсказано 5 пар из 5 реальных |
| Антикодоновый стебель |
5' 38-44 3'
5' 26-32 3'
Всего 7 пар
|
Предсказано 5 пар из 7 реальных |
Предсказано 5 пар из 7 реальных |
| Общее число канонических пар нуклеотидов |
23 пары |
10 пар |
20 пар |
Алгоритм Зукера
Заданная команда без значения параметраа Р
(default P=5)
mfold SEQ=trna.fasta
При увеличенни параметра "P" программа выдавала много структур, которые не оказались лучше первоначальной.
Изображение, полученное с помощью mfold:
Выводы:
Программа einverted, скорее всего, предназначена для работы с ДНК (это видно по замене урацилов на тимины).
На анализе т-Рнк специализируется программа mfold (использующая алгоритм Зуккера), сразу предсказавшая практически
правильную структуру.
Назад