Оптимизация структуры нафталина и азулена с помощью MOPAC
Как и в прошлом задании построим и оптимизируем с помощью MOPAC структуры нафталена и азулена. SMILES молекул представлены ниже:Azulene : C1=CC=C2C=CC=C2C=C1
Napthalene: c1ccc2ccccc2c1
Получились следующие структуры:
| Азулен | Нафталин | |
| До оптимизации | 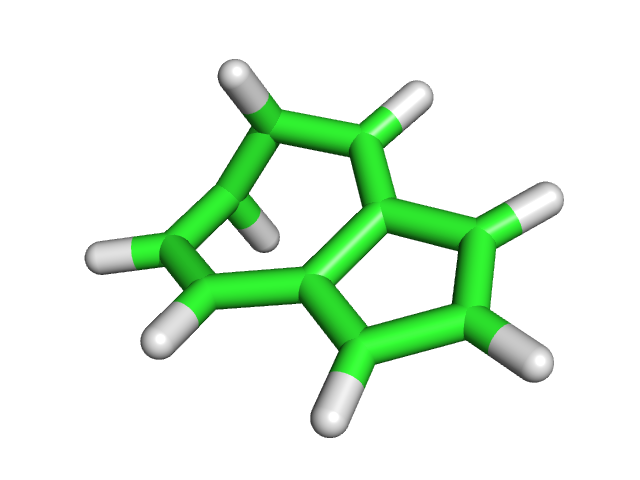 |
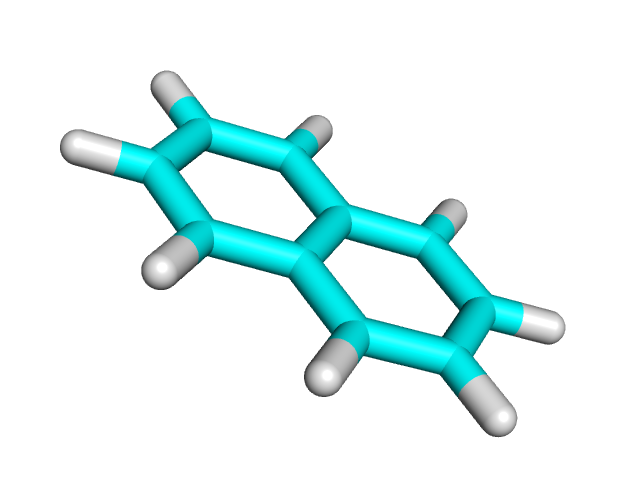 |
| После оптимизации с помощью МОРАС | 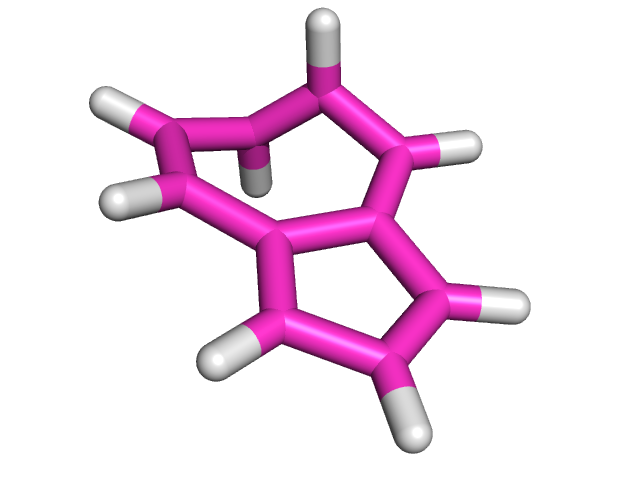 |
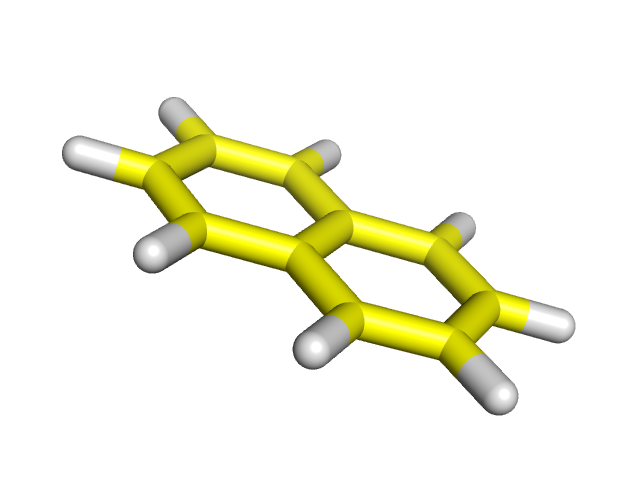 |
| После оптимизации с помощью UFF |  |
obgen az.smi -ff UFF > az_f.mol
Выходные файлы: azu.out, nap.out.
Оптимизация геометрии молекул средствами GAMESS
С помощью babel полученные координаты. были переформатированы в gamin формат:babel -imopout azu.out -ogamin azu.inp
Был вставлен новый заголовок (причем в *.inp необходимы знаки $ и пробелы в начале каждой строки, также не должно быть никаких пустых строк, иначе файл читается неверно):
$CONTRL COORD=CART UNITS=ANGS SCFTYP=RHF RUNTYP=OPTIMIZE $END
$BASIS GBASIS=N31 NGAUSS=6 $end
$system mwords=2 $end
$DATA
Во второй строке заголовка указан базис, с которым будет проводится оптимизация: N31. Новые файлы называются azu.inp и nap.inp. Это и есть входные файлы для оптимизации геометрии средствами GAMESS. Сама оптимизация для обоих файлов была проведена следующим образом (где 1 это количество ядер для расчёта, на текущий момент на kodomo параллельное использование GAMESS не реализовано):
gms nap.inp 1 >& nap.log
При этом в корневой папке появится директория gamess-scratch, в которой будут лежать файлы *.dat.
Расчёт энергии методом Хатри-Фока и методом теории функционала плотности
На основе полученных координат были составлены новые входные файлы для расчёта энергии молекулы методом Хартри-Фока, а также с помощью теории функционала плотности. Для начала log файл gamout были переформатированы в gamin.babel -igamout nap_opt.log -ogamin nap_hf.inp - для расчета методом Хартри-Фока;
cp nap_hf.inp nap_dft.inp - для использования теории функционала плотности (density functional theory, DFT)
Для расчёта по Хартри-Фоку были составлены файлы для нафталена и азулена со следующим заголовком:
$CONTRL COORD=CART UNITS=ANGS SCFTYP=RHF RUNTYP=ENERGY $END
$BASIS GBASIS=N31 NGAUSS=6
POLAR=POPN31 NDFUNC=1 $END
$GUESS GUESS=HUCKEL $END
$system mwords=2 $end
$DATA
Файлы: azu_hf.inp, nap_hf.inp.
В случае теории функционала плотности заголовки файлов были такими:
$CONTRL COORD=CART UNITS=ANGS dfttyp=b3lyp RUNTYP=ENERGY $END
$BASIS GBASIS=N31 NGAUSS=6
POLAR=POPN31 NDFUNC=1 $END
$GUESS GUESS=HUCKEL $END
$system mwords=2 $end
$DATA
Файлы: azu_dft.inp, nap_dft.inp.
Сравнение энергий, полученных методом Хатри-Фока и методом теории функционала плотности
Для каждой из 4 систем был запущен GAMESS аналогично вышеописанному запуску. В выходных файлах были найдены строчки с "TOTAL ENERGY = ". Была составлена таблица энергий. (1 Хартри = 627.509469 ккал/моль)| Вещество | Хартри-Фок | DFT |
| Нафталин | -383.3546610434 | -385.6400107604 |
| Азулен | -383.2824690219 | -385.5857491456 |
| Δ Hartree | 0,072192021 | 0,054261615 |
| Δ kCal/mol | 45,30117708 | 34,04967709 |
Файлы: azu_hf.log, nap_hf.log, azu_dft.log, nap_dft.log.
Оказалось, что метод DFT лучше, так как его результат лучше согласуется с экспериментальным значением энергии изомеризации нафталина в азулен 35.3±2.2 kCal/mol.