Занятие 7&8. Молекулярная динамика биологических
молекул в GROMACS
Цель: ознакомится с возможностями моделирования молекулярной динамики.
В
процессе выполнения задания будем пользоваться пакетом молекулярной динамики
Gromacs.
Общие положения:
Типы
файлов:
·
gro - файл с координатами системы.
·
top - файл с описанием ковалентных и нековалентных
взаимодействий в молекулах.
·
mdp - файл с описанием параметров для работы
молекулярно-механического движка.
·
tpr - файл для молекулярно-механического движка по сути есть
объединение gro, top и mdp.
·
trr, xtc - файл с координатами после расчёта.
Моделирование
перехода ДНК из А в В форму:
1.
Создаём рабочую директорию на диске Н: типа Tereshkova/md
2.
Нам даны файлы, скачаем их в свою рабочую директорию:
·
файл параметров для минимизации энергии em.mdp.
·
файл параметров для "утряски" воды pr.mdp pr.mdp.
·
файл параметров для молекулярной динамики md.mdp.
3.
Зайдём на kodomo через Putty и перейдём в рабочую директорию:
cd Tereshkova/md
4. С помощью программы fiber из пакета 3DNA
построим небольшой дуплекс с последовательностью GATCTA.
Для того, что бы fiber работал надо задать путь и переменную среды:
export X3DNA=/home/preps/golovin/progs/X3DNA export PATH=/home/preps/golovin/progs/X3DNA/bin:${PATH} 5. Скопируем директорию Tereshkova на 172.16.0.140. Зайдём через Putty на
172.16.0.140. Теперь построим файл топологии системы в силовом поле amber99sb
и файл с координатами в формате Gromacs. Предполагается, что структура
дуплекса находится в файле dna.pdb.
pdb2gmx -f dna.pdb -o dna -p dna -ff amber99sb -water tip3p
Получили сообщение об ошибке. Для её устранения
следует удалить 5' фосфаты из структуры дуплекса. Их два на 5' конце каждой
цепи.
Также надо изменить имена нуклеотидов добвавив
D к названию нуклеотида," Т"->"DT" и заменить имя атома
"С5М" на " С7".
6. Сделаем небольшой отступ в ячейке от ДНК:
editconf -f dna.gro -o dna_ec -d 1.5
7. Проведём оптимизацию геометрии системы, что
бы удалить "плохие" контакты в молекуле:
grompp -f em -c dna_ec -p dna -o dna_em -maxwarn 1 mdrun -deffnm dna_em -v
Отметим изменение максимальной силы в ходе
оптимизации геометрии:
начальное и конечное значение максимальной силы
соответственно.
8. Добавим в ячейку молекулы воды:
genbox -cp dna_em -p dna -cs -o dna_s
9. Нейтрализуем заряд системы. Это делаем в два
шага: строим tpr и запускаем genion.
В выводе grompp обратим внимание на информацию
о заряде системы!
grompp -f em -p dna -c dna_s -o dna_s genion -s dna_s -o dna_si -p dna -np X
, где Х это количество положительных ионов
необходимых для нейтрализации заряда системы.
В данном случае х=10.
10. Проведём "утряску" воды:
grompp -f pr -c dna_si -p dna -o dna_pr -maxwarn 1 mdrun -deffnm dna_pr -v
11. Переформатируйте dna_pr.gro и dna_si.gro в pdb формат.
editconf -f my.gro -o my.pdb
И сравним визуально в PyMol изменения в
системах:


Заметное различие между полученными
изображениями. Слева - система до утряски воды (dna_pr.pdb), справа - после (dna_si.pdb).
Видно, что система справа упорядочена, в
отличие от её исходного состояния.
12. Следующий шаг: скопировать файлы на
суперкомпьютер.
Для этого:
·
зайдем на суперкомпьютер и создадим папку с фамилиейи
вернёмся на kodomo
ssh skif mkdir Tereshkova exit
·
скопируем файлы:
scp -r * skif:Tereshkova/
13. Запускаем тестовое моделирование на
суперкомпьютере:
ssh skif cd Tereshkova grompp -f md -c pep_pr -p pep -o pep_md -maxwarn 1 mpirun -np 16 -maxtime 5 -q test /home/golovin/progs/bin/mdrun_mpi -deffnm pep_md -v
Ошибок нет, поэтому можем перейти к основному
моделированию:
mpirun -np 16 -maxtime 1200 /home/golovin/progs/bin/mdrun_mpi -deffnm pep_md -v
Номер задачи: 240958.
Анализ
результатов моделирования перехода А-формы ДНК в
В-форму в воде.
Параметры
моделирования:
·
Силовое поле, используемое при
построении топологии:
amber99sb
·
Заряд системы: 0.
Нейтрализовали заряд системы следющим образом:
genbox -cp dna_em -p dna -cs -o dna_sgrompp -f em -p dna -c dna_s -o dna_s genion -s dna_s -o dna_si -p dna -np 10
Добавив молекулы воды в систему, вычислили её
заряд, в соответствии с чем (заряд системы был равен "-10") было
добавлено 10 катионов.
·
Размер и форма ячейки:
Кубическая ячейка с параметрами
·
Минимизация энергии :
- алгоритм минимизации энергии: integrator
= l-bfgs
- алгоритм использования малого количества
памяти Broyden-Fletcher-;Goldfarb-Shanno (квази-Ньютоновский),
метод аппроксимирует Гауссиановскую матрицу из
предыдущих конфигураций.
-
алгоритм расчёта электростатики и Ван-дер-Ваальсовых взаимодействий: двойное
обрезание (cut-off).
·
Модель, которой описывался растворитель:
растворитель - вода. Модель воды: tip3p.
·
Утряска растворителя:
Для биополимеров укажите параметр, который обуславливает
неподвижность биополимера: define = -DPOSRES.
·
Число шагов
nsteps = 1000000
·
Длина шага
dt = 0.001 ; в pс
·
Алгоритм расчёта электростатики и
Ван-дер-Ваальсовых взаимодействий:
Алгоритм для расчёта электростатики - pme
(суммирование по Эвальду).
Алгоритм для вычисления VdW - cut-off.
·
Алгоритмы термостата и баростата.
Для контроля температуры использовался метод Berendsen.
Контроля давления нет.
·
Основной расчёт МД:
- время моделирования: 4 часа 39 минут 7 секунд
- количество процессоров: 16
- эффективность машстабирования: 100%
- длину траектории : 10000 пс.
- число шагов: 5000000.
- длина шага: 0,002 пс.
- алгоритм интегратора: md
- алгоритм расчёта электростатики и
Ван-дер-Ваальсовых взаимодействий:
·
алгоритм для расчёта электростатики - pme (суммирование по
Эвальду).
·
Ван-дер-Ваальсовых взаимодействий - двойное обрезание
(cut-off).
- алгоритмы термостата и баростата: для
контроля температуры и давления использовался метод Berendsen.
АНАЛИЗ :
1. Любой анализ начинают с визуального анализа
движений молекул. (при вопросе о выводе групп будем выбирать DNA).
trjconv -f dna_md.xtc -s dna_md.tpr -o dna_pbc_1.pdb -skip 20 -pbc mol
Чтобы центрировать ячейку, и иметь возможно
зрительно оценивать результат введём слеующую команду:
trjconv -f dna_md.xtc -s dna_md.tpr -o dna_fit_1.pdb -skip 20 -fit rot+trans
На мой взгляд, переход в В-форму происходит на
8 кадре:
dna_pbc_1.pdb , dna_fit_1.pdb .
Время соответствующего перехода записано в
pdb-файле двумя строчками выше записи о 8 моделе.
Время перехода составляет 1600 пс.
2. Определим среднеквадратичное отклонение в
ходе моделирования. Так как у нас происходит конформационный переход, сначала
рассчитаем отклонение в ходе всей симуляции относительно стартовой структуры:
g_rms -f dna_md.xtc -s dna_md.tpr -o rms_1
Графическое представление:
среднеквадратичное отклонение. rms_1
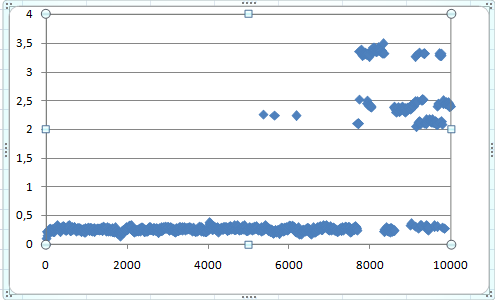
Из графика видно, что среднее квадратичное
отклонение колеблется на уровне 0,25 нм.
Рассмотрим среднеквадратичное отклонение
относительно каждой предыдущей структуры на растоянии 400 кадров. Если ближе к
концу закончился конформационный переход, то отколнение должно уменьшаться:
g_rms -f dna_md.xtc -s dna_md.tpr -o rms_2 -prev 400
Графическое представление: rms_2
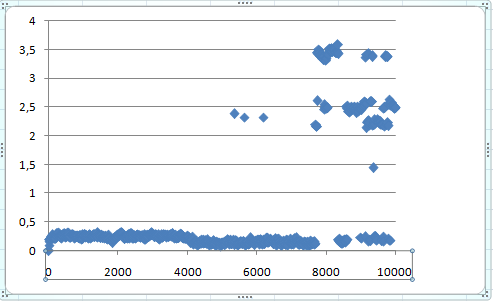
Действительно, к концу среднее квадратичное
отклонение с 0,25 нм снизилось до 0,12-0,15 нм , что свидетельствует об
окончании конформационного перехода.
3. Определим изменение гидрофобной и
гидрофильной поверхности в ходе конформационного перехода:
g_sas -f dna_md.xtc -s dna_md.tpr -o sas_dna.xvg
Графическое представление: гидрофобная
поверхность. sas_dna
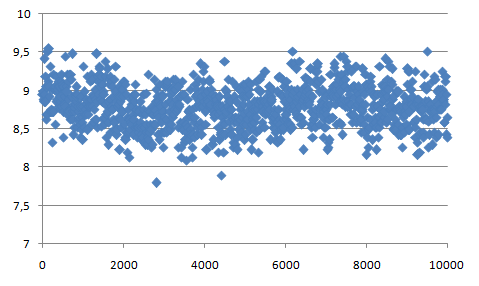
Графическое
представление: гидрофильная поверхность. sas_dna
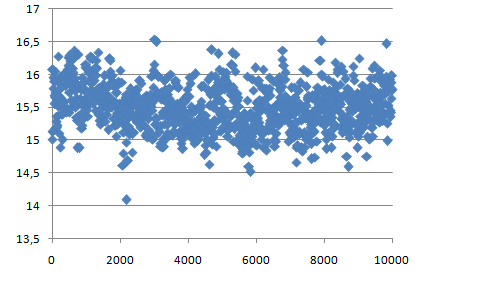
Площадь гидрофобной и гидрофильной поверхности
в процессе конформационного перехода остаются практически без изменений.
4. Традиционным анализом для ДНК является расчёт
колчества образуемых водородных связей.
Если мы будем исследовать связи между ДНК и
ДНК, то это будут водородные связи между цепями ДНК.
Для конца траектории:
g_hbond -f dna_md.xtc -s dna_md.tpr -num hbond_dna
Графическое представление: водородные
связи ДНК-ДНК. hbond_dna
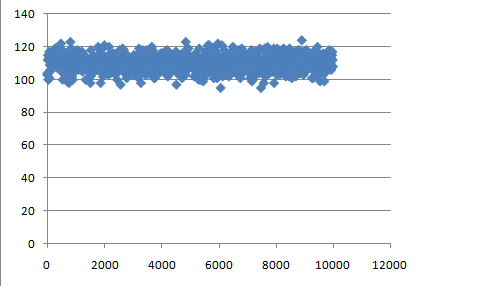
Количество водородных связей, образуемых с
водой, колеблется в пределах от 100 до 120.
Не менее интересно будет изучить количество
вдородных связей ДНК-Вода:
g_hbond -f dna_md.xtc -s dna_md.tpr -num hbond_dna_sol
Графическое
представление: водородные связи ДНК-вода. hbond_dna_sol
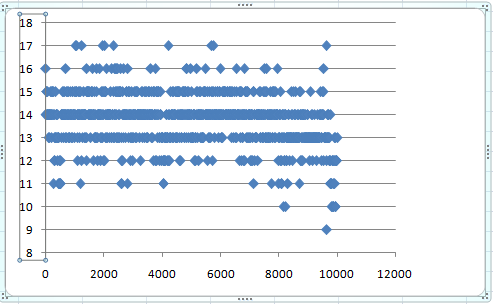
Известно, что водородных связей в дуплексе
должно быть 14. Из графика видно, что, действительно, в процессе перехода
количество водородных связей не сильно изменяется. Диапозон изменений:13-15
связей.
Логично, что не должно происходить каких-либо
существенных изменений в количестве связей данного типа, так как гидрофильная и
гидрофобная поверхности остаются без существенных изменений в процессе
перехода.