1. Подготовка файлов координат и топологии.
На основе gro файла с 38 молекулами этана с прошлого занятия был создан индекс файл, в котором будет группа из одной молекулы этана.
make_ndx -f box_38.gro -o 1.ndx(После запуска команды появляется приглашение к вводу. Для ознакомления с помощью надо нажать кнопку "h" + enter. Далее был выбран остаток номер 1 -> enter -> появилась новая группа). Для создания gro файла с одной молекулой и задания ячейки была введена следующая команда:
editconf -f box_38.gro -o et1.gro -n 1.ndx editconf -f et1.gro -o et.gro -d 2 -c
- последней командой мы задаем ячейку и располагаем молекулу по центру
(При запуске ediconf надо выбрать номер, соответствующий группе из одной молекулы).
В результате был получен файл et.gro.
Также я исправила файл топологии et.top из прошлого задания:
в разделе [ molecules ] надо было просто изменить количество молекул этана (вместо 38-ми 1).
2. Разные параметры контроля температуры.
be.mdp - метод Берендсена для контроля температуры.
vr.mdp - метод "Velocity rescale" для контроля температуры.
nh.mdp - метод Нуза-Хувера для контроля температуры.
an.mdp - метод Андерсена для контроля температуры.
sd.mdp - метод стохастической молекулярной динамики.
Скрипт по работе с 5ю системами одновременно: смотреть.
3. Разбор скрипта.
Входные файлы были построены для молекулярно-динамического движка mdrun с помощью gromp:
grompp -f ${i}.mdp -c et.gro -p et.top -o et_${i}.tpr
(гдеi: be,vr,nh,an,sd - см. выше список mdp файлов)
Для каждого из 5 полученных tpr файлов запускается mdrun.
mdrun -deffnm et_${i} -v -nt 1
Для визуального анализа необходимо конвертировать каждую из 5 систем в pdb:
trjconv -f et_${i}.trr -s et_${i}.tpr -o et_${i}.pdb
Полученные pdb-файлы для дальнейшего просмотра в PyMOL:
- et_be.pdb
- et_an.pdb
- et_nh.pdb
- et_sd.pdb
- et_vr.pdb
Наблюдения, сделанные на основе визуального анализа:
1) В методе Берендсена в начале видны небольшие колебания и вращение молекулы, но достаточно быстро скорость вращения начинает увеличиваться, а амплитуда колебаний затихать. Такая система выглядит довольно неправдоподобно.
2) В методе Андерсена видны небольшие колебания по длинам связей и валентным углам, однако молекула при этом не вращается. Возможно, такое состояние характерно для достаточно низких температур, когда молекулы образуют кристалл.
3) В методе Нуза-Хувера длина связи С-С в молекуле этана практически не изменяется (по ркайней мере, визуально), можно наблюдать лишь вращение. Хотя в основном молекула находится в более выгодной заторможенной конформации, иногда можно заметить заслонённую.
4) В методе стохастической молекулярной динамики трудно оценить по какому принципу молекула движется, т.к. она очень быстро перемещается, меняя свое направление то ли случайно, то ли при столкновении с другими молекулами, которые просто здесь не представлены.
5) Метод "Velocity rescale" и метод Нуза-Хувера весьма похожи друг на друга, разница лишь в том, что в первом методе вращение по связи меньше, а амплитуда колебаний больше. Невыгодные конформации, близкие к заслонённой, также видны достаточно редко.
Для сравнения потенциальной энергии связи и кинетической энергии для каждой из 5 систем:
g_energy -f et_${i}.edr -o et_${i}_en.xvg
На этом месте работа скрипта закончилась.
4. Графики изменения энергий.
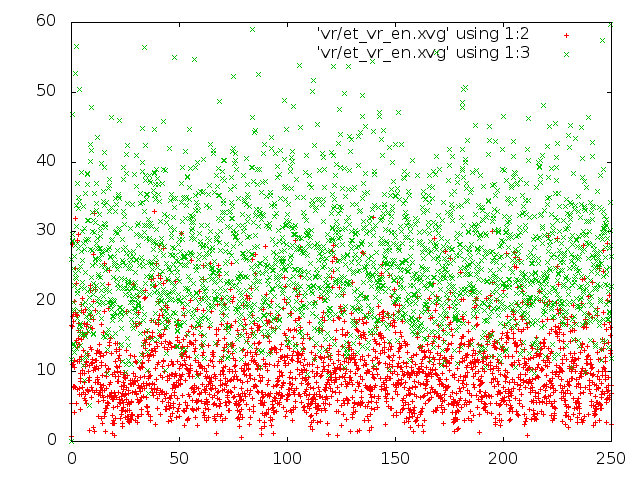
С помощью Gnuplot были построены графики изменения энергий (рекомендуемый вид - dot-plot). Зеленым цветом выделена кинетическая энергия, и зеленым потенциальная.
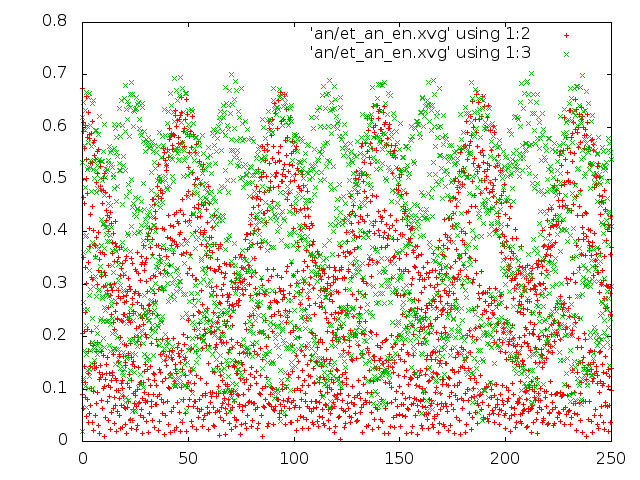
1) В методе Андерсена значения потенциальной и кинетической энергий примерно одинаковые и очень маленькие. Поэтому не верится, что такой термостат хорошо подходит для передачи молекуле необходимой энергии для того, чтобы она попала в глобальный минимум;
2) В методе Берендсена потенциальная энергия довольно быстро уменьшается, а вот кинетическая выходит на плато. Значения кинетической энергии выше, чем в методе Андерсена, но все равно еще достаточно невелики;
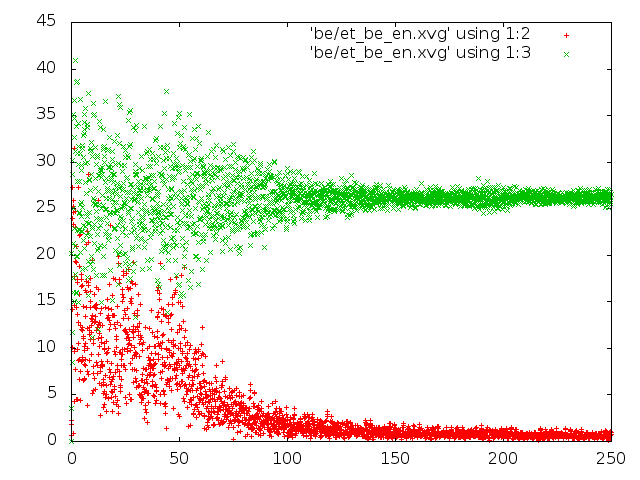
3) В методе Нуза-Хувера можно увидеть единичные достаточно высокие значения кинетической энергии.
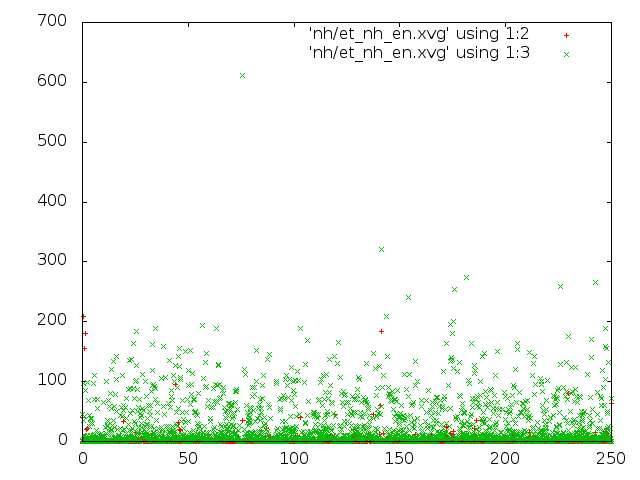
4) В методе стохастической молекулярной динамики видны средние, примерно постоянные, значения кинетической энергии;
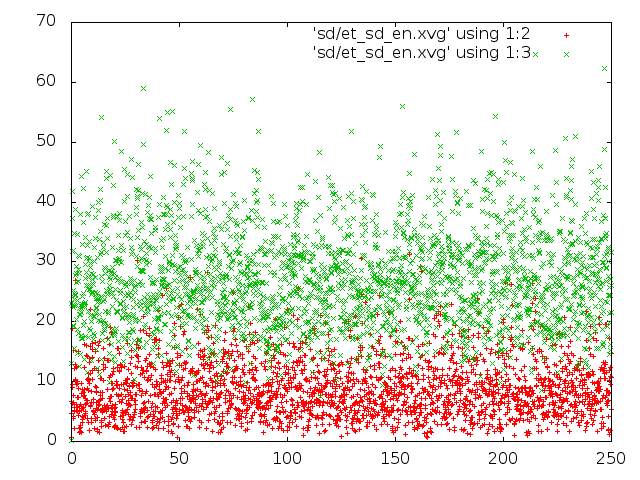
5) В методе "Velocity rescale" ситуация сходна с тем, что происходит в методе стохастической молекулярной динамики.
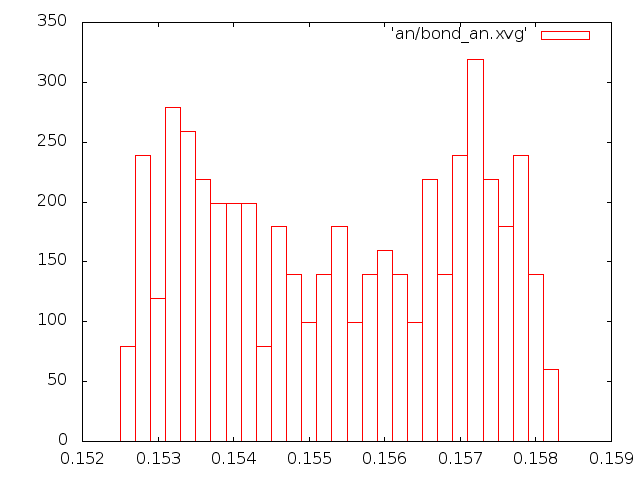
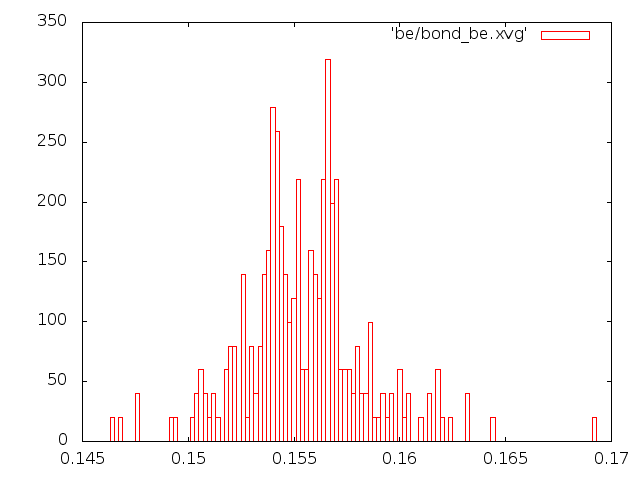
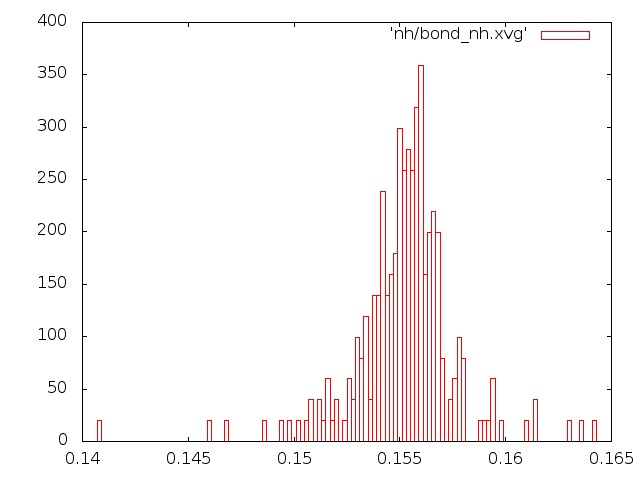
Графики распределения длин связей. Для анализа распределения длины связи С-С за время моделирования в текстовом редакторе был создан индекс-файл b.ndx с одной связью. Его содержимое:
[ b ] 1 2
После этого была запущена утилита по анализу связей g_bond:
g_bond -f et_${i}.trr -s et_${i}.tpr -o bond_${i}.xvg -n b.ndx
Графики распределения длин связей были также построены в Gnuplot:
1) Метод Андерсена
2) Метод Берендсена
3) Метод Нуза-Хувера
4) Метод стохастической молекулярной динамики
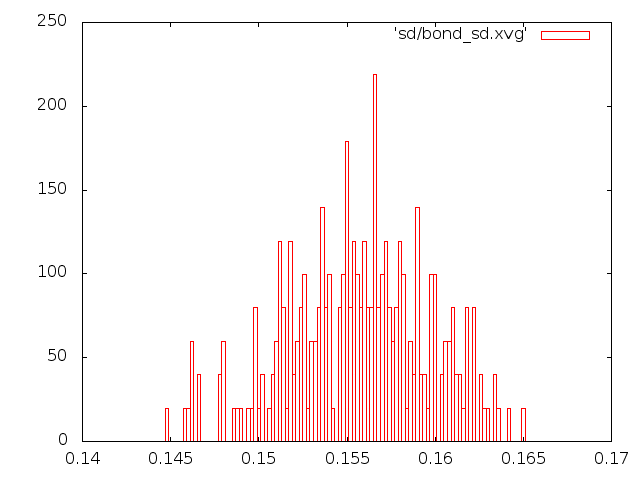
5) Метод "Velocity rescale"
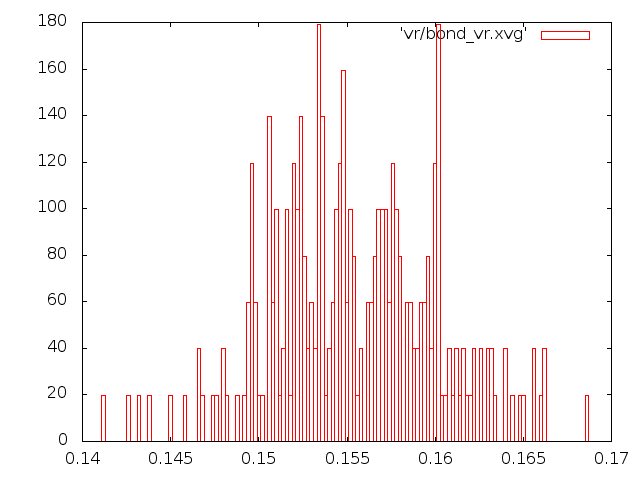
Распределение Больцмана.
Форма распределения Больцмана:
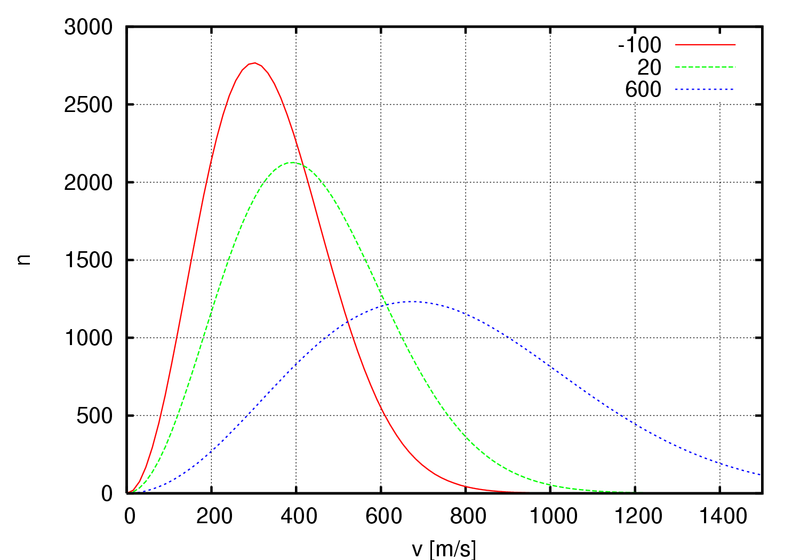
Этому распределению совершенно не соответствуют графики методов Андерсена и Берендсена, а вот с графиками методов стохастической молекулярной динамики и "Velocity rescale" соответствие неплохое.
Исходя из всех полученных наблюдений, можно сказать, что наиболее вероятны 2 метода: стохастической молекулярной динамики и "Velocity rescale". Однако в то же время визуальное представление метода стохастической молекулярной динамики в Pymol довольно сложно интерпретировать, по крайней мере, в данном конкретном случае. Поэтому, по моему мнению, метод, который позволяет наиболее реалистично поддерживать температуру в системе, - "Velocity rescale".