Задание: поэтапное освоение возможностей Mopac как пакета для оптимизации структуры молекул и расчёта некоторых свойств.
1. Оптимизация структуры порфирина
Начнём работу с установки переменных:
export PATH=${PATH}:/home/preps/golovin/progs/bin
export MOPAC_LICENSE=/home/preps/golovin/progs/bin
Взяв аннотацию SMILES о молекуле порфирина, создаём текстовый файл 1.smi,
где сначала строки - SMILES порфирина, а через несколько пробелов просто porphyrin.
На основе этого описания c помощью программы из babel можно построить 3D структуру порфирина (1.mol):
obgen 1.smi > 1.mol
Теперь необходимо с помощью babel переформатировать координаты в mol формате во входной файл для Mopac
(c помощью -xk задаем параметризацию типа pm6 ):
babel -ipdb 1.pdb -omop 1_opt.mop -xk "PM6"
Для полученного файла запустим пакет Mopac:
MOPAC2009.exe 1_opt.mop
Переформатируем результат в pdb (1_opt.pdb):
babel -imopout 1_opt.out -opdb 1_opt.pdb
Проведем оптимизацию с параметризацией AM1:
babel -ipdb myfile.pdb -omop 1_opt.mop -xk "AM1"
Получили следующий результат:
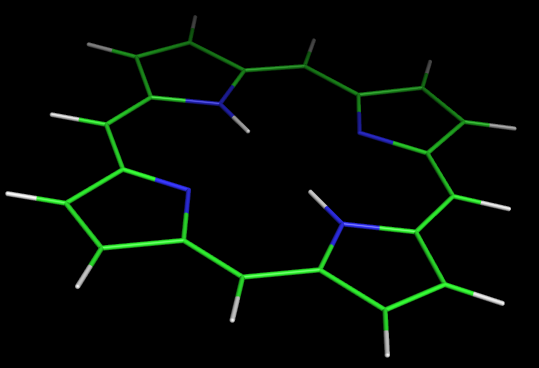
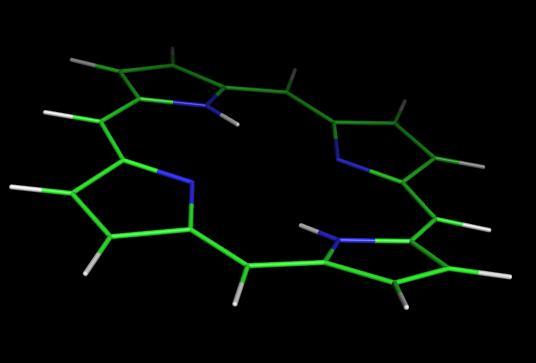
Сравнив полученные структуры порфирина, несложно заметить, что с помощью пакета MOPAC мы получили плоскую структуру, каковой на самом деле и является молекула порфирина.
Сравнение "профилей" полученных структур: (слева - результат OBGEN; справа - результат MOPAC):

Таким образом, с помощью MOPAC можно получить верную структуру.
2. Расчет некоторых свойств полученной структуры порфирина
Рассчитаем возбужденные состояния порфирина и на основе этих данных оценим спектр поглощения молекулы.
Для расчёта возбуждённых состояний используем ранее полученный файл mop.
Преобразуем этот файл, дописав в конец следующие строки:
пустую строку cis c.i.=4 meci oldgeo some description
Запускаем MOPAC для рассчетов:
MOPAC2009.exe 1_opt_spectr.mop
Нас интересует записи в конце полученного файла (1_opt_spectr.mop) со значениями энергий для электронных переходов:
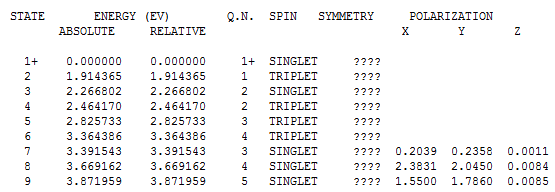
Рассчитаем длины волн, при которых происходят эти переходы:

Согласно литературным данным, спектр поглощения порфиринов включает два диапазона - 400-410 нм и 580-660 нм.
2. Оптимизация структуры парабензохинона
Для данной молекулы необходимо провести выше указанные процедуры как с помощью OBGEN, так и MOPAC.
Получили следующие изображения (слева-OBGEN, справа-MOPAC):
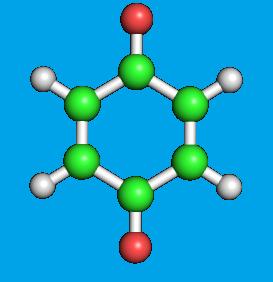
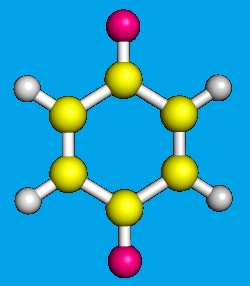
Теперь, определим геометрию дианиона этой молекулы. Для этого в первую строчку mop файла добавляем слово CHARGE=-2.
Заттем явным способом указываем, что на атомах кислорода должен находиться отрицательный заряд. Пример:
PM6 CHARGE=-2 gg O(-) 0.98570 1 0.00130 1 -0.43680 1 C 2.16830 1 0.00680 1 -0.12400 1 ...........
Получаем изображение дианиона:
При наложении структур получаем следующее изображение:
(на картинке: малиновый кислород и зелёный углерод - дианион; бирюзовый углерод и красный кислород - структура парабензохинона без изменений)
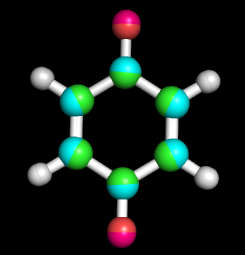
За счёт того, что в молекуле дианиона связи С-О - одинарные, видно, что кислороды в этой молекуле находятся на более длинном расстоянии.