Построим выравнивание последовательности из структуры ID: 1lmp и белка LYSC_COLLI. Используем программу Clustal.
Полученное выравнивание сохраняем в формате PIR.
Теперь модифицируем файл выравнивания:
Переименуем последовательность в файле выравнивания, как в примере:
| БЫЛО | СТАЛО |
| >P1;UNIPROT|P37712|LYSC_CAMDR | >P1;SEQ |
| >P1;1LMP__|PDBID|CHAIN|SEQUENCE | >P1;1LMP |
sequence:ХХХХХ::::::: 0.00: 0.00эта строчка описывает входные параметры последовательности для modeller.
После имени последовательности белка-образца добавляем:
structureX:1lmp_now.ent:1 :A: 132 :A:undefined:undefined:-1.00:-1.00эта строчка описывает, какой файл содержит структуру белка с этой последовательностью, номера первой и последней аминокислот в структуре, идентификатор цепи и т.д.
В конце каждой последовательности добавляем символы:
/.Символ "/" означает конец цепи белка. Точка указывает на то, что имеется один лиганд (если бы было два лиганда стояли бы две точки).
Модифицированное выравнивание
Теперь необходимо модифицировать файл со структурой. Для этого удалим всю воду из структуры (в текстовом редакторе), всем атомам лиганда присвоим один и тот же номер "остатка" (MODELLER считает, что один лиганд = один остаток) и модифицируем имена атомов каждого остатка, добавив в конец буквы A, B, C. Смысл операции в том, что атомы остатка 130 имели индекс А, атомы остатка 131 имели индекс В и т.д. . После модификации имен атомов измените номера остатков на 130.
Пример:
| БЫЛО | СТАЛО |
| HETATM 1014 O7 NAG 130 | HETATM 1014 O7A NAG 130 |
| HETATM 1015 C1 NAG 131 | HETATM 1015 C1B NAG 130 |
Полученный файл
Теперь перейдем к созданию управляющего скрипта
У нас есть следующая заготовка:
from modeller.automodel import *
class mymodel(automodel):
def special_restraints(self, aln):
rsr = self.restraints
for ids in (('OD1:98:A', 'O6A:131:A'),
('N:65:A', 'O7B:132:A'),
('OD2:73:A', 'O1C:133:A')):
atoms = [self.atoms[i] for i in ids]
rsr.add(forms.upper_bound(group=physical.upper_distance,
feature=features.distance(*atoms), mean=3.5, stdev=0.1))
env = environ()
env.io.hetatm = True
a = mymodel(env, alnfile='test1.ali', knowns=('1lmp'), sequence='seq')
a.starting_model = 1
a.ending_model = 5
a.make()
В скрипте указано:- что нужно использовать стандартные валентные углы в полипептидной цепи (строчка 4)
что дополнительно нужно сохранять взаимное расположение определенных пар атомов (3.5 ангстрема);
В данном случае трех атомов белка, образующих водородные связи с тремя атомами лиганда - строчки 5-7 с ID пар атомов; параметры взаимного расположения атомов пары описаны в строчке 9-10. 3 точки могут однозначно расположить сложную структуру в пространстве, поэтому мы выбираем водородные связи как источник данных точек.
- что ковалентные связи в гетероатомах нужно вычислять по расстояниям между атомами (так же, как это делает Rasmol), строчка 12
- что имя файла с выравниванием и имена последовательностей образца и моделируемого белка, строчка 13 (а имя файла со структурой содержится в выравнивании)
- что число и номера моделей, которые нужно построить (в данном примере 5 моделей), строки 14-15
- что пора строить модель строчка 16
В скрипте необходимо отредактировать строчки, в которых указаны какие водородные связи белка с лигандом должны быть В БУДУЩЕЙ МОДЕЛИ. Номера остатков и имена нужных атомов определим по выравниванию и тому, какие водородные связи имеются в образце. Критерий водородной связи: расстояние менее 3.5 ангстрем между азотом или кислородом белка с подходящими атомами лиганда.
Если в моделируемом белке число остатков не совпадает с числом остатков в белке-образце, то номера "остатков" лиганда изменятся.
ВНИМАНИЕ. В скриптовом языке (на основе python ) важно с какого столбца начинается информация. Необходимо соблюдать предложенный в заготовке синтаксис.
Полученный управляющий скрипт
Запустим исполнение скрипта командой:
mod9v7 myscript &
Просмотрим полученные модели.
 |  |
 | 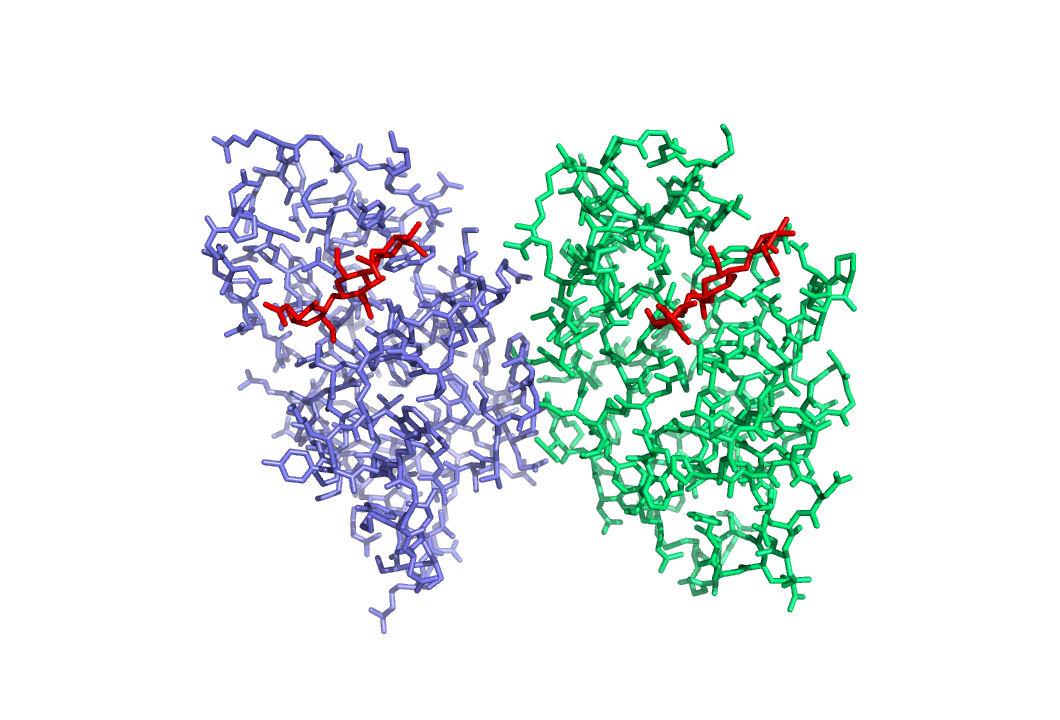 |
 | 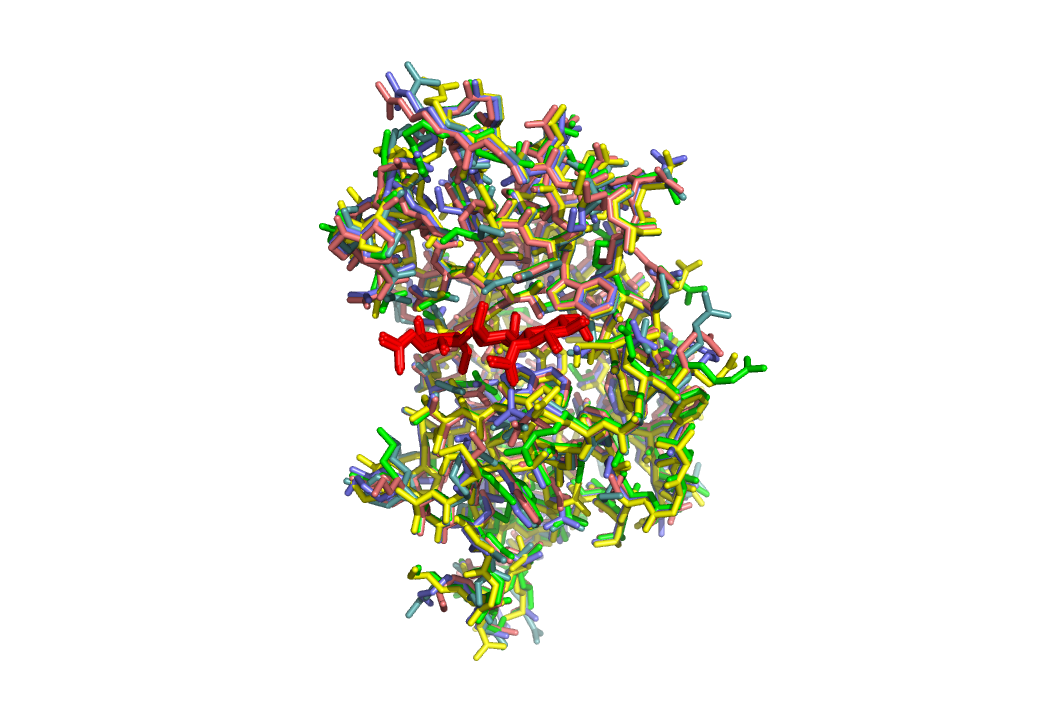 |
Как видим они лишь немного различаются по расположению боковых групп.
Проверим качество моделей и выберем лучшую. Инструменты для оценки качества структуры можно найти в веб интерфейсе WHATIF .