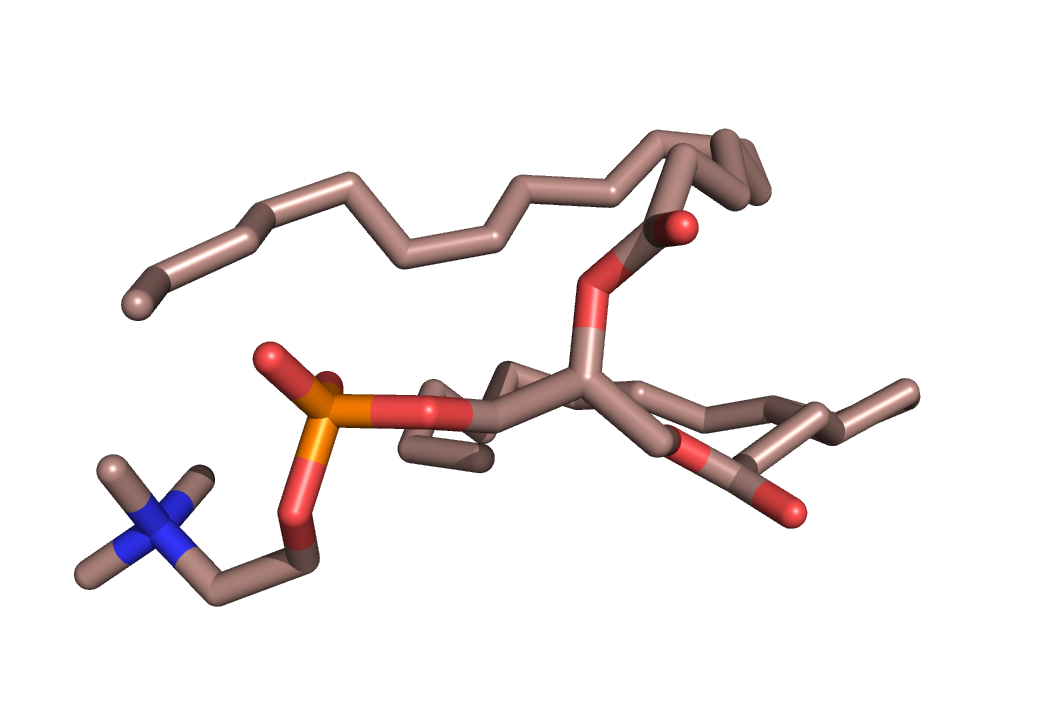
Цель: ознакомится с возможностями моделирования молекулярной динамики.
Полезные ссылки:Уроки по работе с GROMACS находятся здесь. Сведения о работе с Gnuplot см. здесь. Введения о скриптовании в Bash здесь. Ведения о awk здесь
Вся работа по расчётам будет проходить на kodomo через терминал putty, а для работы с графическим выводом Gnuplot понадобится Xming. В этом занятии мы будем пользоваться пакетом молекулярной динамики Gromacs. Это программное обеспечение распространяется под лицензией GPL, т.е. пользователь может скачать исходный код и свободен его изменять по своему усмотрению
Типы файлов:
1. gro - файл с координатами системы. 2. top - файл с описанием ковалентных и нековалентных взаимодействий в молекулах. 3. mdp - файл с описанием параметров для работы молекулярно-механического движка. 4. tpr - файл для молекулярно-механического движка по сути есть объединение gro, top и mdp. 5. trr, xtc - файл с координатами после расчёта.
Основные программы из пакета, которые будут использованы на занятии:
Программы запускаются в командной строке Linux, флаги запуска программ начинаются с -, например -f. Как правило после флага следует либо имя файла, либо значение параметра.
1. editconf - манипуляция форматом координат и самими координатами.
editconf -f my.gro -o my.pdb2. genbox - наполнение ячейки растворителем.
genbox -cp my.gro -cs mysolvent.gro -p my.top -o my_solvated.gro3. genion - утилита для замены n молекул растворителя на ионы.
genion -s my.tpr -np 10 -p my.top -o my_ions.gro -np это добавить 10 положительно заряженых ионов4. grompp - объединение и проверка gro, top и mdp в tpr.
grompp -f my.mdp -c my.gro -p my.top5. mdrun - молеклярно-механический движок. На входе принимает tpr файл.
mdrun -deffnm my.tpr
здесь параметр -deffnm означает, что выходные файлы будут называться как и входной файл, только с другими расширениями.
Всю работу проводим на компьютере 172.16.0.140
После запуска задачи нахождение её в очереди можно посмотреть с помощью: tasks -o
дополнительной топологии для липида DPPC, dppc.itp.
параметры для липидов lipid.itp.
координаты одного липида dppc.gro.
Файл-заготовка топологии системы b.top.
файл параметров для минимизации энергии em.mdp.
файл параметров для "утряски" воды pr.mdp pr.mdp.
файл параметров для молекулярной динамики md.mdp
genconf -f dppc.gro -o b_64.gro -nbox 4 4
| |
editconf -f b_64.gro -o b_ec -d 0.5
grompp -f em -c b_ec -p b -o b_em -maxwarn 2 mdrun -deffnm b_em -v
В ходе оптимизации геометрии изменяется значение силы действующей на молекулы, значения начальное и конечное максимальной силы 4.37970e+05 и 6.4541919e+02 .
genbox -cp b_em -p b -cs spc216 -o b_s
grompp -f pr -c b_s -p b -o b_pr -maxwarn 1 mdrun -deffnm b_pr -v
Переформатируем b_pr.gro и b_s.gro в pdb файлы:
 | 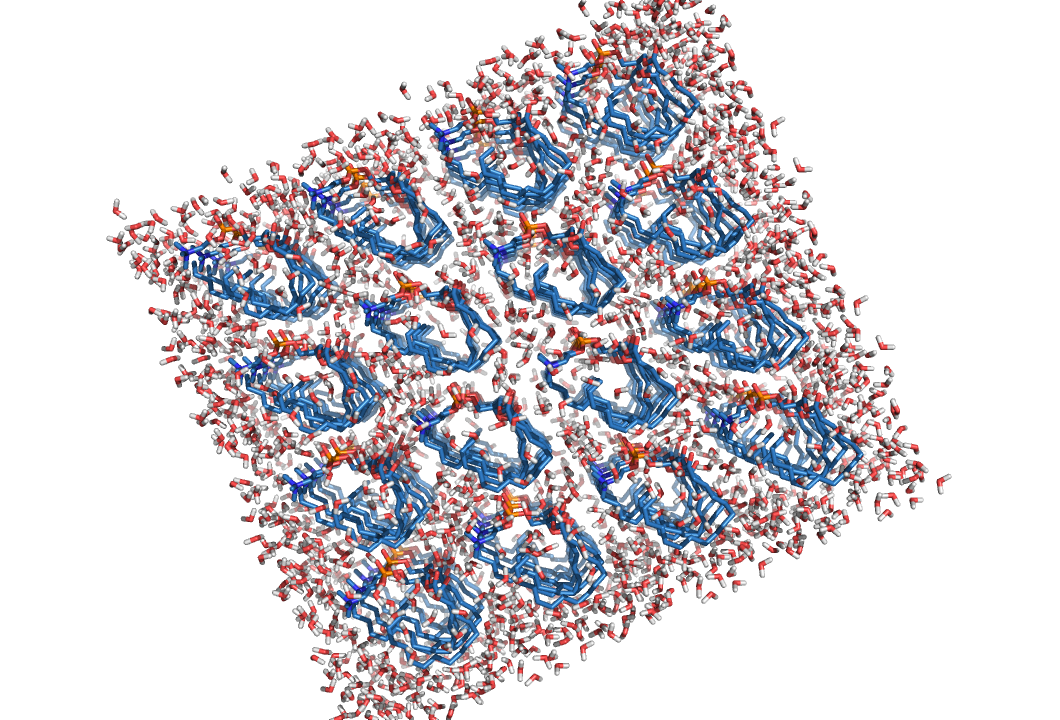 |
ssh skif mkdir Piskunova exit
cd .. scp -r md/* skif:Piskunova/
ssh skif cd Piskunova grompp -f md -c b_pr -p b -o b_md -maxwarn 1 mpirun -np 16 -q test -maxtime 5 /home/golovin/progs/bin/mdrun_mpi -deffnm b_md -v
Необходимо запомнить номер задачи.Просмотреть ход счёта можно в файле mdrun_mpi.out-номер.
less mdrun_mpi.out-номер Нажмите shift и . для перехода в конец файла.
Если файл не содержит ошибок, то переходим дальше:
mpirun -np 16 -maxtime 1200 /home/golovin/progs/bin/mdrun_mpi -deffnm b_md -v less mdrun_mpi.out-.... Нажмите shift и . для перехода в конец файла.