Узнаем аннотацию порфирина в виде SMILES на сайта. Создадим файл 1.smi , в нем сначала идут строки SMILES порфирина, а через несколько пробелов просто porphyrin.
На основе этого описания c помощью программы из babel построим 3D структуру порфирина:
obgen 1.smi > 1.molРассмотрим полученную структуру в PyMol и удалим ненужные водороды, получим файл.
|
|
|
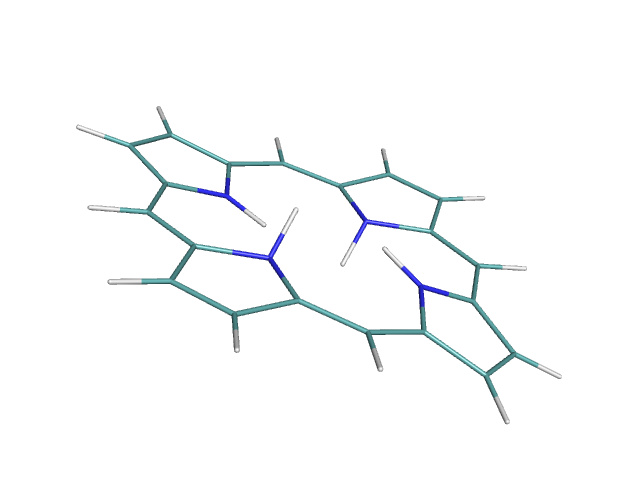
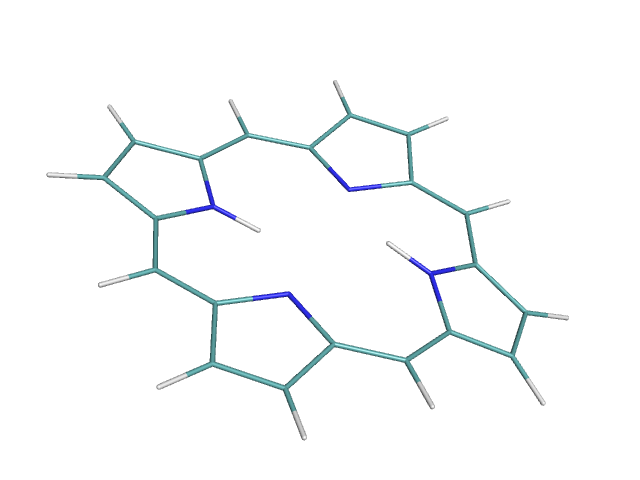
babel -ipdb 1.pdb -omop 1_opt.mop -xk "PM6"Запустим Mopac для файла:
MOPAC2009.exe 1_opt.mopИзучим файл вывода out. Для сравнение переформатируем результат в pdb:
babel -imopout 1_opt.out -opdb 1_opt.pdbПолучили:
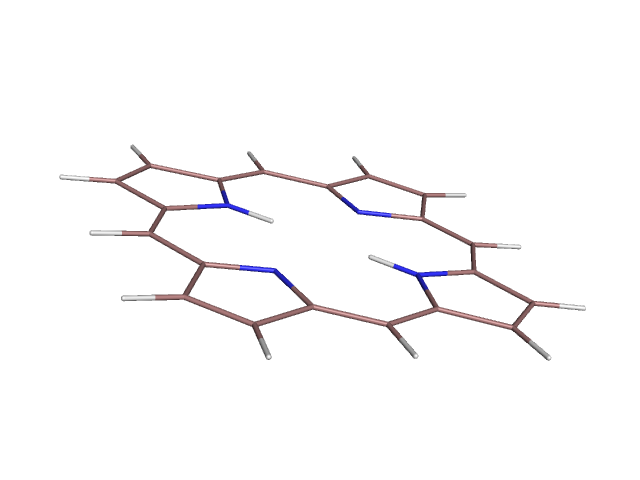
|
|
|
пустую строку cis c.i.=4 meci oldgeo some descriptionЗапустим Mopac:
MOPAC2009.exe 1_opt_spectr.mop
В конце файла находятся значения энергий для электронных переходов:
STATE ENERGY (EV) Q.N. SPIN SYMMETRY POLARIZATION
ABSOLUTE RELATIVE X Y Z
1+ 0.000000 0.000000 1+ SINGLET ????
2 1.913312 1.913312 1 TRIPLET ????
3 2.266014 2.266014 2 SINGLET ????
4 2.463186 2.463186 2 TRIPLET ????
5 2.823915 2.823915 3 TRIPLET ????
6 3.362161 3.362161 4 TRIPLET ????
7 3.389757 3.389757 3 SINGLET ???? 0.2031 0.2347 0.0010
8 3.669242 3.669242 4 SINGLET ???? 2.3899 2.0438 0.0085
9 3.871323 3.871323 5 SINGLET ???? 1.5461 1.7992 0.0084
Рассчитаем длины волн, при которых происходят эти переходы ( Е=h*c/длина волны ):Энергия (эВ) Длина волны (нм) 1,913312 648,913002 2,266014 547,910575 2,463186 504,0516769 2,823915 439,6637412 3,362161 369,2782808 3,389757 366,2719876 3,669242 338,3731664 3,871323 320,7102672
|
|
|
Теперь, определим геометрию дианиона этой молекулы. Для этого в первую строчку mop файла добавляем слово CHARGE=-2. Потом явным способом указываем на каких атомах должен находиться отрицательный заряд. Пример:
PM6 CHARGE=-2 gg O(-) 0.98570 1 0.00130 1 -0.43680 1 C 2.16830 1 0.00680 1 -0.12400 1 ...........
Работаем с файлом для Mopac. В итоге получаем файл-pdb. Сравним полученные структуры:
|
дианион |
просто |
Сравнение:
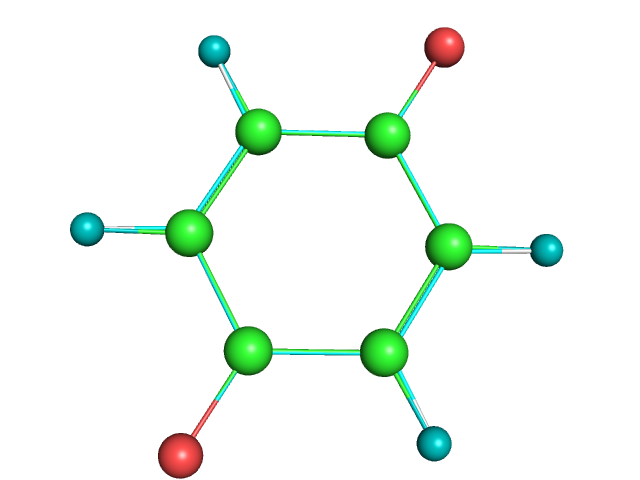
Видим, что структуры немного отличаются по расположению связей. Так же атомы кислорода выглядят эллипсоидами- в связи с тем, что у дианиона эти атомы находятся чуть дальше, чем у незаряженной молекулы.
babel -p 7.4 -ipdb test.pdb -opdb test_opt.pdbЗатем к файлу добавим вручную в файл атом магния где-то между гамма-фосфором и СА аспартата (можно просто скопировать атом фосфора и поменять имя атома на Mg и поменять координаты как среднее арифметическое между координатами гамма-фосфора и СА аспартата):
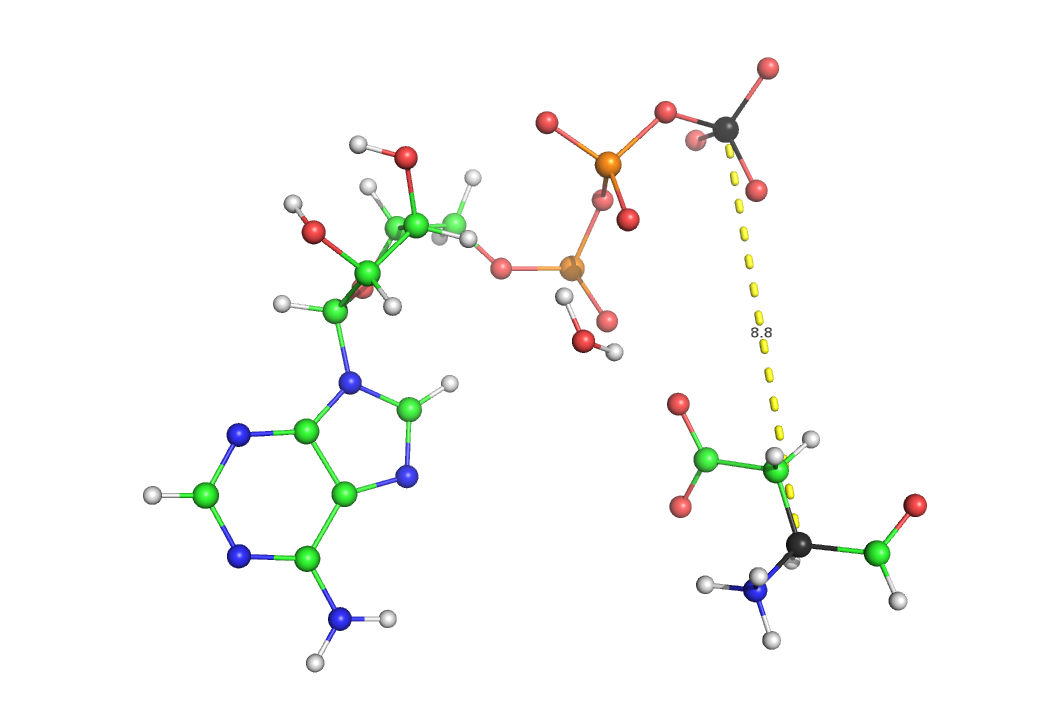
babel -ipdb testdop.pdb -omop td.mop babel -ipdb testdop.pdb -oxyz td.xyzСледующим этапом будет запрет движения для всех атомов кроме гамма фосфата, воды и магния. Этот шаг нам нужен для того, чтобы потом легко восстановить эту конформацию в белке. Для "заморозки атомов" надо в mop файле поменять 1 после координаты на 0. Пример:
PM6 CHARGE=-3 test N -33.26800 0 25.03000 0 4.67600 0 .......Получим файл для работы.
Как удобно определить каким атомам надо разрешить двигаться? Откройте xyz файл в PyMol и отобразите labels как atoms id. Номер строчки с этим атомом в mop файле будет id+3. Таким образом установите у нужных атомов 1 по всем координатам - это атомы в строках 14, 43, 44, 64 и 65.Проведем оптимизацию (PM6) с помощью MOPAC, получим файл.
В итоге получили следующие изображение:
|
|
|
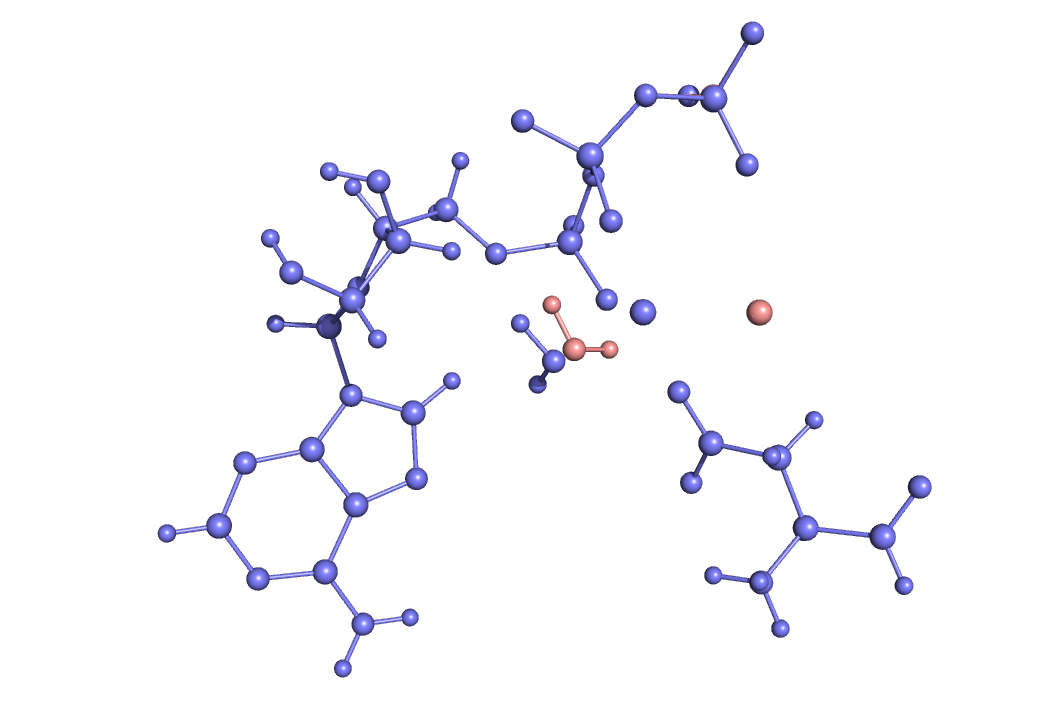
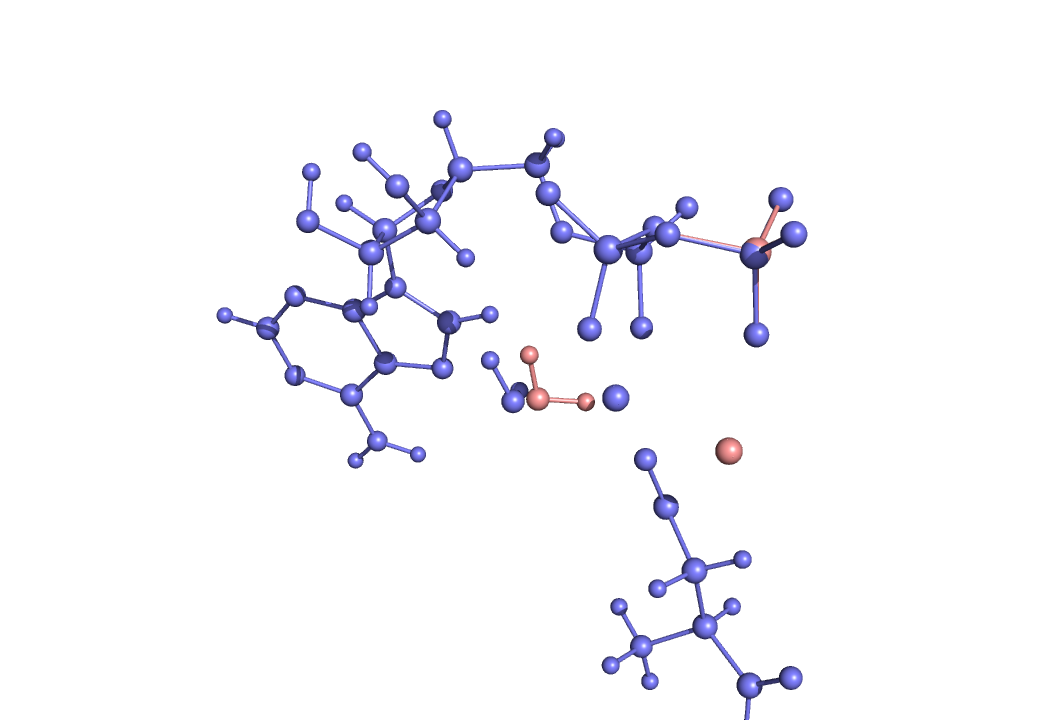
Видим, что произошел поворот воды, она немного подвинулась, так же изменилось немного положение гамма-фосфора, сильно изменилась позиция иона магния. Атом магния располагается более симметрично между водой, фосфатом и са аспартата.
Если рассмотреть pdb-запись 3pp1, то можно заметить:
|
|
|
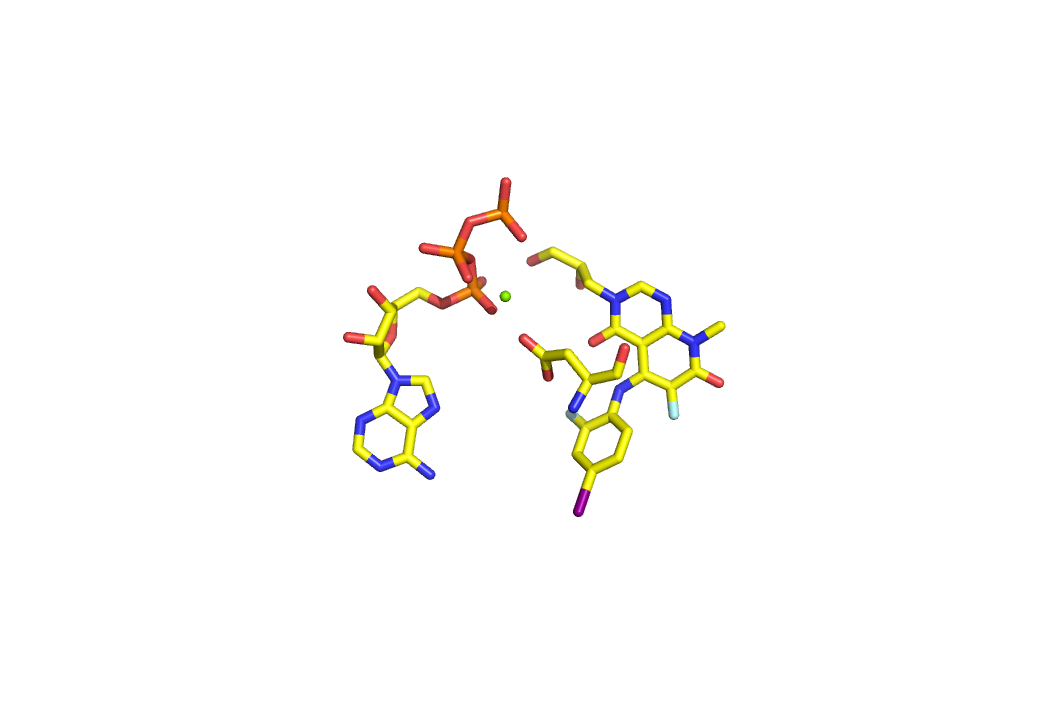
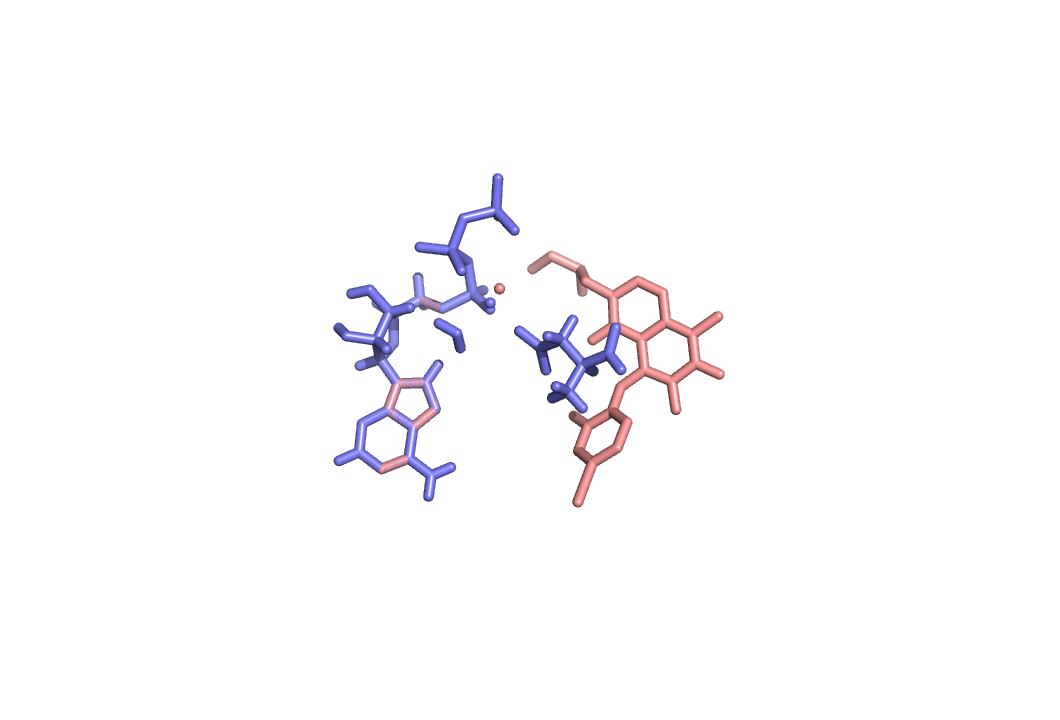
Что расположение этой структуры в реальной молекуле весьма похоже. Но все таки атом магния не на своем месте, конформация остатка аспартата не такая же, остаток фосфата так же под другим углом...
Впринципе, можно сказать, что MOPAC вполне нормально справился, тк не учитывались заряды.