Запускаем retree.exe. Программа выдает следующее сообщение:
Tree Rearrangement, version 3.69
Settings for this run:
U Initial tree (arbitrary, user, specify)? User tree from tree file
N Format to write out trees (PHYLIP, Nexus, XML)? PHYLIP
0 Graphics type (IBM PC, ANSI, none)? IBM PC
W Width of terminal screen, of plotting area? 80, 80
L Number of lines on screen? 24
Are these settings correct? (type Y or the letter for one to change)
Нажмем кнопку Y. Программа выдала укоренение и спросила следующую команду, выбираем M ("Midpoint root the tree").
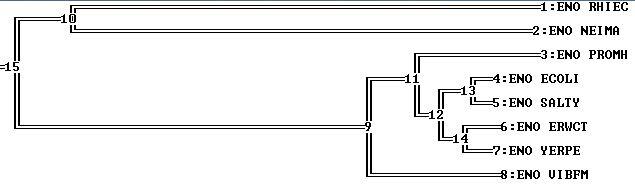
Укоренение прошло по ветви (RHIEC,NEIMA) против ((PROMH,((ECOLI,SALTY),(ERWCT,YERPE))),VIBFM).
Такое укоренение совпадает с алгоритмом UPGMA.
Дерево, построенное методом максимальной бережливости, укоренить нельзя, так как данный метод не предполагает существования молекулярных часов и не выдает длин ветвей. Дерево же , построенное методом UPGMA, не имеет смысл укоренять, так как метод UPGMA выдает уже укорененное дерево.
В качестве аутгруппы используем белок того же семейства (ENO) из сенной палочки (Bacillus subtilis, BACSU).
Для того, чтобы добавить последовательность белка из сенной палочки, используем команду:
seqret sw:eno_bacsu stdout >> eno.fasta
Получили файл с последовательностями. Затем построим выравнивание наших последовательностей с помощью muscle:
muscle -in eno.fasta -out enob_aligned.fasta
Получили файл с выровненными последовательностями. В этом файле от названий последовательностей оставляем мнемонику видов, и подаем его на вход программе fprotpars.
Для работы с этой программой используем команду:
fprotpars -sequence enob_aligned.fasta -outfile enob.fprotpars
На выходе получили изображение дерева и скобочную модель.
Обработаем полученное дерево с помощью программы retree, используем действие "select an Outgroup"(O) и указываем номер BACSU.
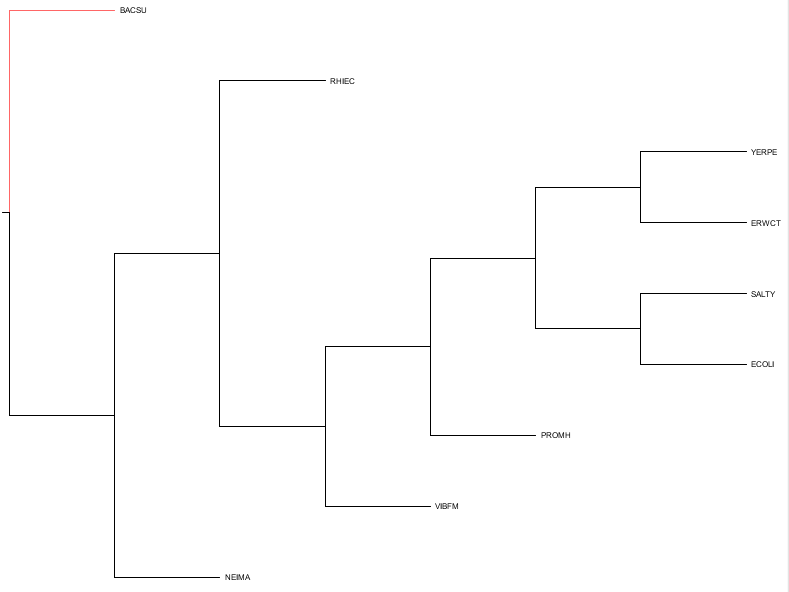
Деревья, полученные с помощью алгоритмов UPGMA и Neighbor - Joining, совпадают с полученным деревом, но все они содержат отличия от истинного дерева. Они обьединяют в одну кладу YERPE и ERWCT. Такое укоренение отличается от укоренения в среднюю точку, оно разносит по разные стороны укоренения NEIMA и все остальные организмы. Такое укоренение, так же отличается от "правильного".
Проведем бутстрэп-анализ филогении, используя программу fprotpars. Для этого создадим 100 бутстрэп-реплик выравнивания белков протеобактерий программой fseqboot.
По полученным репликам получим деревья программой fprotpars (для этого просто подадим выходной файл программы fseqboot на вход программе fprotpars). Получили скобочные версии деревьев .
Создадим из полученных деревьев полученных деревьев единое дерево по принципу "расширенного большинства" (extended majority rule tree). Для этого файл с деревьями, выданный программой fprotpars, подадим на вход программе fconsense.
Получили файл с изображением дерева .
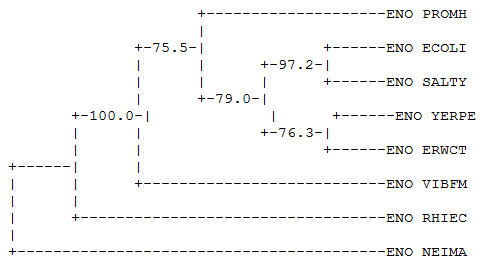
Полученное дерево полностью повторяет полученные ранее деревья. Ошибка содержится в ветви, которая получила поддержку 76.3. Однако в тоже время ветвь с меньшей поддержкой - 75.5 - правдива.