Пространственное выравнивание и совмещение
Команда align PyMOL
С помощью команды align PyMOL были совмещены соответствующие домены цепей A двух структур: 1HY0 и 1I0A. Доменная структура молекул была получена с помощью pDomains, используя метод CATH. В обоих молекулах было найдено по три домена. (сессия PyMOL)
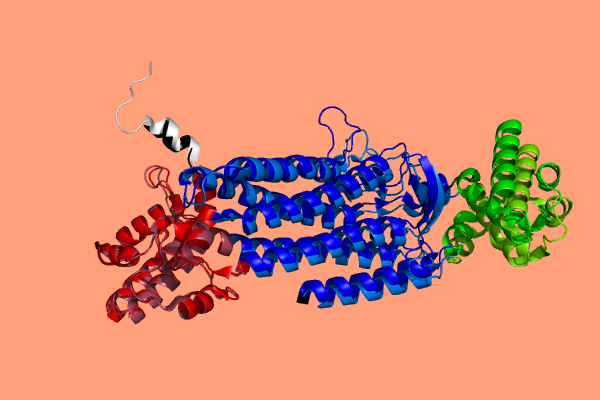
Выравнивание по первому домену (rmsd=0.497)
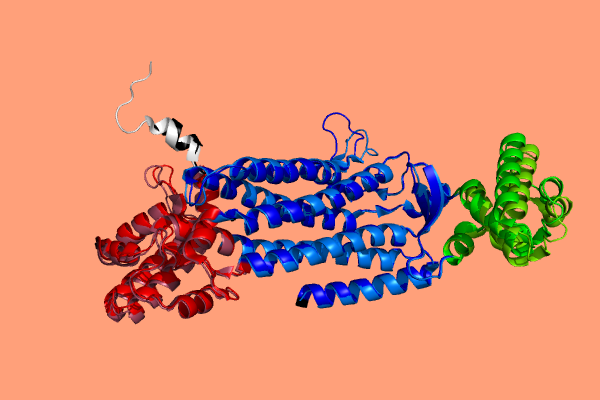
Выравнивание по второму домену (rmsd=0.531)
Выравнивание по третьему домену (rmsd=0.557)
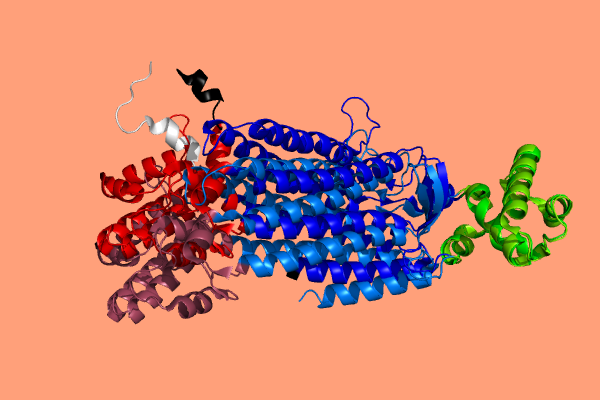
(На всех изображениях первый домен красный, второй - синий, третий - зеленый; 1HY0 - черная и ярко красная, зеленая и синяя, 1I0A - белая и красная, зеленая и синяя с непривычным оттенком)
На изображениях видно, что домены выравниваются хорошо (маленькие rmsd и просто визуально), но остальная структура при этом не совпадает, т.е. одна молекула немного изогнута, по сравнению с другой и домены располагаются немного по-разному друг относительно друга. По совмещению можно сказать, что команда align хорошо справляется, выравнивая очень похожие структурные фрагменты.
PDBeFOLD, Geometrical core
С помощью PDBeFOLD было построенно структурное выравнивание цепей A двух записей банка PDB: 1AKH и 1W0T (rmsd=2.62). (результат в fasta формате)
Командой pair_fit PyMOL было построенно совмещение двух цепей по CA атомам геометрического ядра. (сессия PyMOL)
Совмещение по геометрическому ядру (rmsd=0.865)
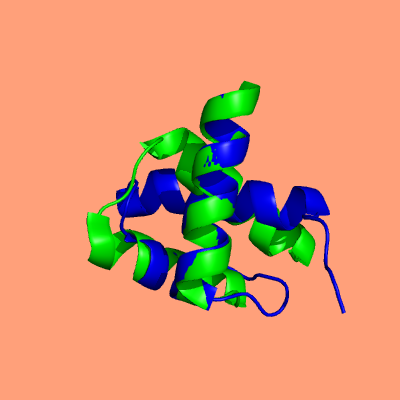
Совмещение по полным цепям с помощью команды align (rmsd=3.088)
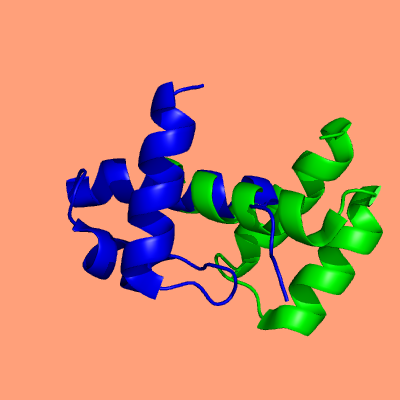
На изображениях видно, что совмещение по геометрическому ядру дает приемлемый результат даже в достаточно сложном случае, тогда как команда align в данном случае не справилась, выравнивая не очень похожие структурно и совсем не похожие по последовательности цепи.

2010
©