Программы obgen, babel и Mopac
Задание 1.
Получил из интернета SMILES порфирина.
(файл .smi)
С помощью программы obgen постороил 3D структуру порфирина:
obgen porphyrin.smi > porphyrin.mol
(файл .mol)
Открыл полученную структуру в PyMOL и удалил лишние водороды.
(файл .pdb)
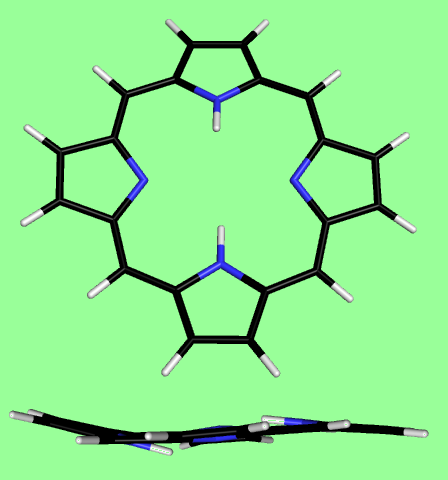
Полученная структура
С помощью babel переформатировал файл в входной формат Mopac с параметризацией PM6:
babel -ipdb porphyrin_h.pdb -omop porphyrin_opt.mop -xk "PM6"
(файл .mop)
Запустил Mopac:
MOPAC2009.exe porphyrin_opt.mop
(файл .out)
С помощью babel переформатировал выходной файл в pdb формат:
babel -imopout porphyrin_opt.out -opdb porphyrin_opt.pdb
(файл .pdb)
Оптимизированная структура
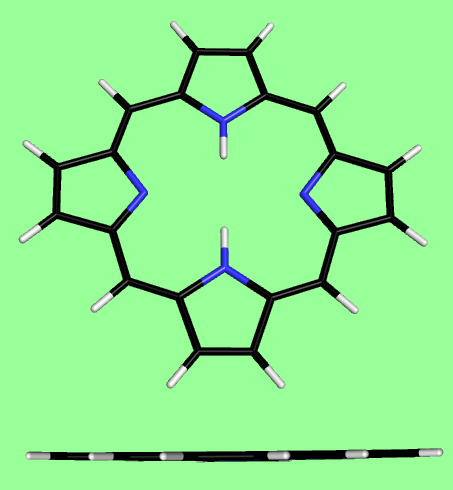
Структура полученная с помощью obgen далека от реальности - в ней присутствуют два водорода на азотах, которые лишают цикл ароматичности, поэтому структура не плоская. После удаления лишних водородов вручную и оптимизации структуры с помощью Mopac, используя параметризацию PM6, структура стала плоской, что больше похоже на реальность. Также была проведена оптимизация с помощью Mopac с параметризацией AM1, результат отличается от предыдущего совсем незначительно.
(файл .pdb)
Вывод: для предстказания пространственной структуры даже не очень сложных ароматических соединений не достаточно программы obgen, следует оптимизировать структуру с помощь Mopac.
Задание 2.
Для расчета возбужденных состояний порфирина отредактировал входной файл Mopac.
(файл .mop)
Запустил Mopac:
MOPAC2009.exe porphyrin_opt_spectr.mop
(файл .out)
Нашел информацию о энергии возбужденных состояний:
STATE ENERGY (EV) Q.N. SPIN SYMMETRY POLARIZATION
ABSOLUTE RELATIVE X Y Z
1+ 0.000000 0.000000 1+ SINGLET ????
2 1.913312 1.913312 1 TRIPLET ????
3 2.266014 2.266014 2 SINGLET ????
4 2.463186 2.463186 2 TRIPLET ????
5 2.823915 2.823915 3 TRIPLET ????
6 3.362161 3.362161 4 TRIPLET ????
7 3.389757 3.389757 3 SINGLET ???? 0.2031 0.2347 0.0010
8 3.669242 3.669242 4 SINGLET ???? 2.3899 2.0438 0.0085
9 3.871323 3.871323 5 SINGLET ???? 1.5461 1.7992 0.0084
The "+" symbol indicates the root used.
С помощью Excel посчитал длины волн, при которых происходят данные переходы:
λ=c*h*109/δE*e, где с - скорость всета, h - постоянная Планка, e - заряд электрона
δE (EV) λ (nm)
1,913312 647,9989361
2,266014 547,1387822
2,463186 503,3416642
2,823915 439,0444261
3,362161 368,7581113
3,389757 365,7560528
3,669242 337,8965302
3,871323 320,2585112
Задание 3.
Для молекулы парабензохинона O=C1C=CC(=O)C=C1 определил геометрию как с помощью obgen
(файл .smi) (файл .mol) (файл .pdb)
так и Мopac.
(файл .mop) (файл .out) (файл .pdb)
Потом определил структуру дианиона молекулы с помощью Mopac.
(файл .mop) (файл .out) (файл .pdb)
Структуры, полученные для бензохинона с помощью Mopac и obgen, отличаются незначительно:
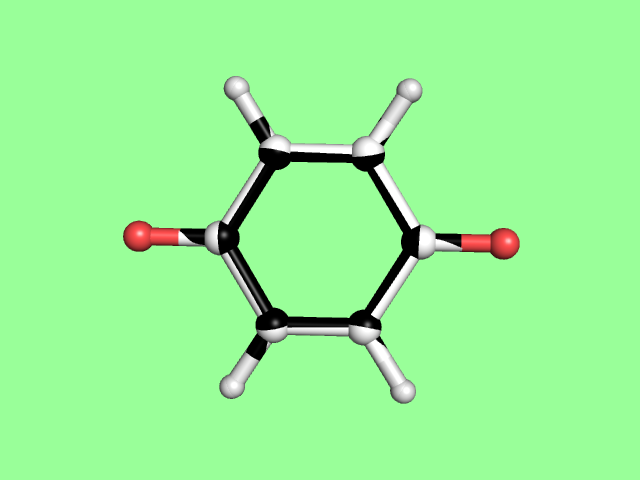
Черным цветом выделена структура obgen, белым - Mopac.
Структура дианиона заметно отличаются от структур незаряженных молекул:
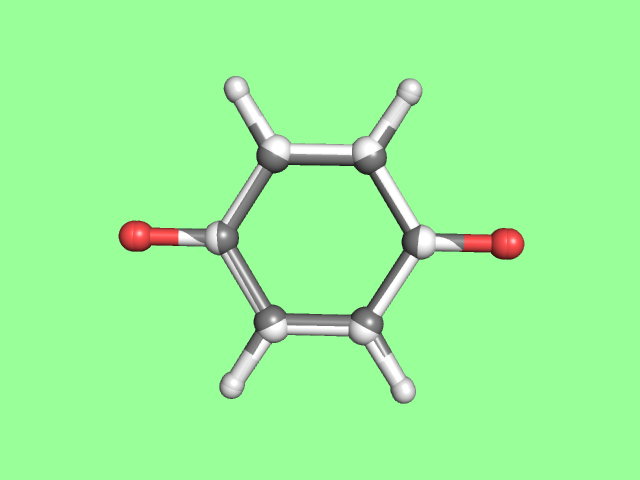
Серым цветом выделена структура дианиона, белым - структура Mopac незаряженной молекулы.
На картинке видно, что заметно увеличилось растояние между кислородами (5.3Å в незаряженной молекуле и 5.5Å в дианионе), т.е. увеличились длины связей C=O. Объяснить такое изменение можно тем, что дианион бензохинона (а точнее бензохинола) представляет собой ароматическую систему, и связи С-O скорее одинарные, чем двойные, а значит они длиннее. Для сравнения, для молекулы бензохинола растояние между кислородами 5.5Å, как и в случае полученного дианиона.
Структура бензохинола, полученная с помощью obgen
(файл .smi) (файл .mol) (файл .pdb)
и Mopac.
(файл .mop) (файл .out) (файл .pdb)
Задание 4.
С помощью babel добавил в данную структуру водороды при рН=7.0:
babel -ipdb test.pdb -opdb test_h.pdb -p 7.0
Потом вручную добавил ион Mg2+ между γ-фосфором АТФ и Сα аспартата:
(исходный файл .pdb) (полученный файл .pdb)
С помощью babel преобразовал pdb файл в формат mop, а потом вручную проставил заряды и запреты на движение для всех атомов, кроме воды, магния и γ-фосфата АТФ.
(файл .mop)
Запустил Mopac, полученную структуру сравнил с записью банка PDB 3PP1.
(файл .out) (файл .pdb)
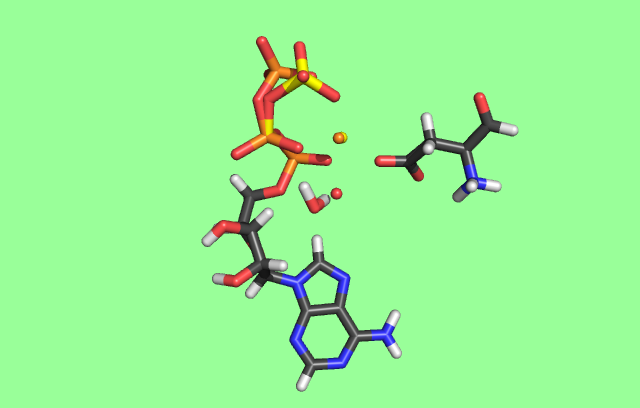
Оранжевым цветом выделены фосфаты и магний из структуры из 3PP1, желтым - из структуры Mopac.,
Исходная структура полностью идентична фрагменту 3PP1, но без магния, а структура, полученная с помощью Mopac содержит ион магния почти в том же месте, что и 3PP1, но изменены положения атомов γ-фосфата АТФ и воды, таким образом Mopac восстановил положение магния, но изменил положение некоторых групп.

2011
©