Занятие 9. Гомологичное моделирование комплекса белка с лигандом.
Цель работы - ознакомиться с возможностями гомологичного моделирования комплекса белка с лигандом с помощью пакета Modeller.
Работать будем с лизоцимом зеленой морской черепахи (LYSC_CHEMY).
Используя известную структуру лизоцима форели (запись PDB 1LMP) как образец,
нужно построить модель комплекса человеческого лизоцима с лигандом.
- Выравнивание 1LMP и LYSC_CHEMY.
С помощью программы ClustalW
построили выравнивание последовательностей белка 1LMP и LLYSC_CHEMY:
смотреть.
- Модификация файла выравнивания.
Теперь надо подготовить файл выравнивания для работы с Modeller. Для этого переименовали последовательности на ">P1;1lmp" и ">P1;seq",
а после имени последовательности моделируемого белка добавили строчку:
sequence:ХХХХХ::::::: 0.00: 0.00
Эта строчка описывает входные параметры последовательности для modeller.
После имени последовательности белка-образца добавили строчку:
structureX:1lmp_now.ent:1 :A: 130 :A:undefined:undefined:-1.00:-1.00
Эта строчка описывает, какой файл содержит структуру белка с этой последовательностью
(1lmp_now.ent), номера первой и последней аминокислот в структуре, идентификатор цепи и т.д.
В конце каждой последовательности добавили символы:
/.
Этот символ означает конец цепи белка. Точка указывает на то, что имеется один лиганд. При этом оставляем строчку со звездочкой (*).
- Модификация файла со структурой.
Удалили из структуры всю воду, всем атомам лиганда присвоили один и тот же номер "остатка" (130), одно и то же имя "остатка" (NAG),
модифицируем имена атомов каждого остатка, добавив в конец буквы A, B и C (чтобы у каждого атома было разное имя).
Смысл операции в том, что атомы остатка 130 имели индекс А, атомы остатка 131 имели индекс В и т.д.
1lmp_now.ent.
- Скрипт.
Управляющий скрипт lysc_chemy.py:
from modeller.automodel import *
class mymodel(automodel):
def special_restraints(self, aln):
rsr = self.restraints
for ids in (('OD2:102:A', 'O6A:131:B'),
('ND2:104:A', 'O7A:131:B'),
('N:110:A', 'O6C:131:B')):
atoms = [self.atoms[i] for i in ids]
rsr.add(forms.upper_bound(group=physical.upper_distance,
feature=features.distance(*atoms), mean=3.5, stdev=0.1))
env = environ()
env.io.hetatm = True
a = mymodel(env, alnfile='lysc_aligned.pir', knowns=('1lmp'), sequence='seq')
a.starting_model = 1
a.ending_model = 5
a.make()
В нем указано:
-
что нужно использовать стандартные валентные углы в полипептидной цепи (строчка 4);
что дополнительно нужно сохранять взаимное расположение определенных пар атомов (3.5 ангстрема) -
в данном случае трех атомов белка, образующих водородные связи с тремя атомами лиганда - строчки 5-7 с ID пар атомов;
параметры взаимного расположения атомов пары описаны в строчке 9-10.
3 точки могут однозначно расположить сложную структуру в пространстве, поэтому мы выбираем водородные связи как источник данных точек.
-
что ковалентные связи в гетероатомах нужно вычислять по расстояниям между атомами, строчка 12;
-
имя файла с выравниванием и имена последовательностей образца и моделируемого белка, строчка 13 (а имя файла со структурой содержится в выравнивании);
-
число и номера моделей, которые нужно построить (в данном примере 5 моделей), строки 14-15;
-
что пора строить модель строчка 16.
Критерий водородной связи: расстояние менее 3.5 ангстрем между азотом или кислородом белка с подходящими атомами лиганда.
Запуск скрипта:
mod9v7 lysc_chemy &
- Полученные модели.
В результате работы скрипта были получены структуры 5 моделей:
LYSC.B99990001.pdb
LYSC.B99990002.pdb
LYSC.B99990003.pdb
LYSC.B99990004.pdb
LYSC.B99990005.pdb
Изображение со структурами моделей, наложенными друг на друга:
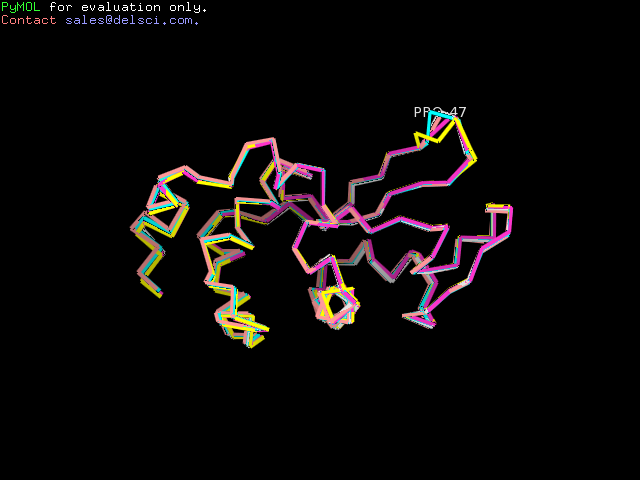
Как видно, моделей практически не отличаются друг от друга. Небольшое расхождение есть только в одной петле (правый верх рисунка), где пролин 47
в разных моделях находися в разном положении.
- Выбор лучшей модели.
Проверим качество моделей. Для этого будем использовать сервис WHATIF
Первая модель
Structure Z-scores, positive is better than average:
1st generation packing quality : -2.225
2nd generation packing quality : -2.551
Ramachandran plot appearance : 0.226
chi-1/chi-2 rotamer normality : -1.310
Backbone conformation : -1.923
RMS Z-scores, should be close to 1.0:
Bond lengths : 0.917
Bond angles : 1.286
Omega angle restraints : 0.793
Side chain planarity : 0.324 (tight)
Improper dihedral distribution : 1.007
Inside/Outside distribution : 0.922
Вторая модель
Structure Z-scores, positive is better than average:
1st generation packing quality : -2.056
2nd generation packing quality : -2.479
Ramachandran plot appearance : 0.376
chi-1/chi-2 rotamer normality : -2.121
Backbone conformation : -1.714
RMS Z-scores, should be close to 1.0:
Bond lengths : 0.930
Bond angles : 1.291
Omega angle restraints : 0.803
Side chain planarity : 0.262 (tight)
Improper dihedral distribution : 1.148
Inside/Outside distribution : 0.935
Третья модель
Structure Z-scores, positive is better than average:
1st generation packing quality : -2.007
2nd generation packing quality : -2.079
Ramachandran plot appearance : 0.228
chi-1/chi-2 rotamer normality : -1.647
Backbone conformation : -1.940
RMS Z-scores, should be close to 1.0:
Bond lengths : 0.958
Bond angles : 1.306
Omega angle restraints : 0.813
Side chain planarity : 0.248 (tight)
Improper dihedral distribution : 1.133
Inside/Outside distribution : 0.915
Четвертая модель
Structure Z-scores, positive is better than average:
1st generation packing quality : -2.116
2nd generation packing quality : -2.342
Ramachandran plot appearance : 0.240
chi-1/chi-2 rotamer normality : -1.986
Backbone conformation : -2.311
RMS Z-scores, should be close to 1.0:
Bond lengths : 0.929
Bond angles : 1.302
Omega angle restraints : 0.726 (tight)
Side chain planarity : 0.392 (tight)
Improper dihedral distribution : 1.084
Inside/Outside distribution : 0.924
Пятая модель
Structure Z-scores, positive is better than average:
1st generation packing quality : -2.146
2nd generation packing quality : -2.050
Ramachandran plot appearance : -0.018
chi-1/chi-2 rotamer normality : -2.324
Backbone conformation : -1.951
RMS Z-scores, should be close to 1.0:
Bond lengths : 0.926
Bond angles : 1.308
Omega angle restraints : 0.852
Side chain planarity : 0.280 (tight)
Improper dihedral distribution : 1.082
Inside/Outside distribution : 0.920
Сравним параметры Ramachandran plot appearance (карта Рамачандрана) и Bond lengths (длины связей). Самый лучший показатель в первом параметре у 2
модели: 0.376 (наиболее положительный), а во втором у 3 модели: 0.958 (наиболее близок к 1). Однако значения параметров для всех моделей достаточно близки
(исключение составляет 5 модель, для которой значение для первого параметра отрицательное, в то время как в других моделях оно положительно), поэтому трудно
выбрать лучшую модель. Для дальнейшего анализа белка LYSC_CHEMY в принципе можно использовать любую модель, кроме, скорее всего, пятой, но наиболее удачным выбором будет 2 или 3 модели, так
как у них лучшие показатели.
|
|