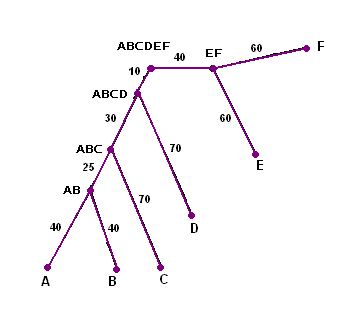
Полученные последовательности, соответствующие листьям дерева, были помещены в виде "выравнивания" в файл fdnaml.fasta. Далее были реконструированы филогенетические деревья с помощью следующих алгоритмов:
Алгоритм максимального правдоподобия (программа fdnaml)
Дерево было получено выполнением программы: fdnaml fdnaml.fasta -ttratio 1 -auto.
+--------B
|
| +------------F
| +-----------4
| +------3 +--------------E
| | |
1------2 +---------------D
| |
| +----------------C
|
+--------A
Алгоритм Neighbor-joining
+--------B
!
! +-----------------C
3------4
! ! +----------------D
! +-------2
! ! +--------------E
! +------------1
! +------------F
!
+--------A
Алгоритм UPGMA
+-----------------A
+--------------1
+------3 +-----------------B
! !
+-------4 +--------------------------------C
! !
--5 +---------------------------------------D
!
! +--------------------------E
+--------------------2
+--------------------------F
Использованные для построения деревьев программы:
fdnadist fdnaml.fasta -ttratio 1 -auto
fneighbor fdnaml.fdnadist -auto (полученный файл был переименован в Neighbor-joining.fneighbor)
fneighbor fdnaml.fdnadist -treetype u -auto
Сравнение деревьев и алгоритмов их построения
| Ветвь дерева |
Исходное дерево |
Дерево, реконструированное алгоритмом максисмального правдопадобия |
Дерево, реконструированное алгоритмом Neighbor-joining |
Дерево, реконструированное алгоритмом UPGMA |
| A |
B |
C |
D |
E |
F |
| |
| . |
. |
* |
* |
* |
* |
+ |
+ |
+ |
+ |
| . |
. |
. |
* |
* |
* |
+ |
+ |
+ |
+ |
| . |
. |
. |
. |
* |
* |
+ |
+ |
+ |
+ |
Из полученной таблицы видно, все деревья, построенные тремя алгоритмами, имеют все ветви исходного дерева.
Если же сравнивать сами алгоритмы, то можно сделать вывод, что только алгоритм UPGMA строит укорененное дерево, что является безусловно наиболее удобным и более информативным результатом.
Деревья, полученные другими двумя алгоритмами можно укоренить искуственно. Это можно сделать двумя способами: укоренение средней точки - "центра тяжести" дерева, либо с помощью метода аутгрупп - выбор гена из достаточно далекого организма и предположение о наиболее вероятной близости узла, в котором ответвляется данная последовательность, к искомому корню.
Также нельзя не заметить принципиальное отличие работы программ, выполнящих алгоритм маскимального правдоподобия и отстальные алгоритмы. Первая программа (алгоритм переборный, символьноориентированный) работает с полным выравниванием полученных мутанатных последовательностей гена. Другие алгоритмы, называемые эвристическими, работают с подсчитанными программой fdnadist попарными эволюционными расстояниями между генами-мутантами, что безусловно является менее информативным, нежели работа с полным выравниванием, поскольку получаемая матрица эволюционных расстояний может соответствовать разным последовательностям.
Алгоритм UPGMA не взвешивает длины ветвей, в итоге строя ультраметрическое дерево. Алгоритм Neighbor-joining, являясь более прогрессивным, взвешивает длины ветвей. Данные особенности алгоритмов хорошо видны по построенным деревьям. Отсюда можно сделать вывод, что алгоритм максимального правдоподобия вполне оправдывает свое название, но его лучше использовать прежде всего для небольшого числа последовательностей. В случае же, когда необходимо как можно быстрей и в то же время правдоподобно представить дерево для большого количества последовательностей, используют алгоритмы UPGMA или Neighbor-joining.
Вернуться на страничку четвертого семестра
© Головкина Мария Сергеевна