Моделирование поведения короткого пептида в формальдегиде.
Проведение моделирования
- Даны файлы:
- Построим файл топологии системы в силовом поле amber99sb и файл с координатами в формате Gromacs.
pdb2gmx -f 2xl1.pdb -o pep -p pep -ff amber99sb -water tip3p
- Сделаем небольшой отступ в ячейке от пептида.
- Проведём оптимизацию геометрии системы, что бы удалить "плохие" контакты в молекуле.
-
Добавим в ячейку молекулы формамида.
- Теперь надо изменить в текстовом редакторе файл тополгии pep.top. После строчки:
-
Нейтрализуем заряд системы. Это делаем в два шага: строим tpr и запускаем genion.
- Проведём "утряску" формамида:
-
Переформатируем pep_pr.gro и pep_si.gro в pdb формат. И сравним визуально в PyMol изменеия в системах.

На картинке слева система до утряски, молекулы строго упорядочены. Справа - после утряски, система выглядит довольно хаотичной.
- Теперь надо скопировать файл на суперкомпьютер. Сначала зайдем на суперкомпьютер и создадим папку с фамилией и вернёмся на kodomo.
- Копируем файлы:
- Запускаем тестовое моделирование на суперкомпьютере.
- Запускаем основное моделирование на суперкомпьютере.
Скачиваем их в рабочую директорию.
editconf -f pep.gro -o pep_ec -d 1.5
grompp -f em -c pep_ec -p pep -o pep_em -maxwarn 1 mdrun -deffnm pep_em -v
В ходе оптимизации геометрии максимальная сила изменялась в пределах порядков 2 и 3. Начальное значение 4.37039e+03, конечное 3.2095328e+02.
genbox -cp pep_em -p pep -cs fam_em.gro -o pep_sВ выводе программы указано, что добавленно 892 молекулы формамида.
; Include forcefield parameters
добавим #include "fam.itp"
; Include forcefield parameters #include "fam.itp"
Добавим количество молекул формамида в запись [ molecules ]
[ molecules ] ; Compound #mols Protein_chain_A 1 FAM 892
grompp -f em -p pep -c pep_s genion -s pep_s -o pep_si -p pep -np 10
где 10 - это количество положительных ионов необходимых для нейтрализации заряда системы. Обратный заряд был указан в выводе grompp
grompp -f pr -c pep_si -p pep -o pep_pr -maxwarn 1 mdrun -deffnm pep_pr -v
ssh skif mkdir Aydarkhanov exit
scp -r * skif:Aydarkhanov/
ssh skif cd Aydarkhanov grompp -f md -c pep_pr -p pep -o pep_md -maxwarn 1 mpirun -np 16 -maxtime 5 -q test /home/golovin/progs/bin/mdrun_mpi -deffnm pep_md -v
Ошибок нет, переходим к основному моделированию.
mpirun -np 16 -maxtime 1200 /home/golovin/progs/bin/mdrun_mpi -deffnm pep_md -v
Номер задачи: 240906. Просмотреть ход счёта можно в файле mdrun_mpi.out-240906.
less mdrun_mpi.out-240906 Shift+. для перехода в конец файла.
Параметры моделирования
- Силовое поле используемое при построении топологии: amber99sb.
- Заряд системы: -10. Причины этого значения (?).
- Размер и форма ячейки: параллелепипед со сторонами (нм) 5.06500, 4.67000, 4.22100.
- Минимизация энергии (файл em.mdp):
- Алогритм минимизации энергии.
integrator = l-bfgs
Квази-Ньютоновский алгоритм в соответствии с методом Бройден-Флетчер-Голдфарб-Шано (BFGS) использования малого количества памяти. - Алгоритм расчёта электростатики и Ван-дер-Ваальсовых взаимодействий.
; Method for doing electrostatics coulombtype = Cut-off
Объединить области отсечения со списком отсеченя соседей (neighborlist cut-off) rlist и отсечения для Кулоновских взаимодействий (Coulomb cut-off) rcoulomb, где rcoulomb ≥ rlist.; Method for doing Van der Waals vdw-type = Cut-off
Объединить области отсечения со списком отсеченя соседей (neighborlist cut-off) rlist и отсечения Ван-дер-Ваальсовых взаимодействий (VdW cut-off) rvdw, где rvdw ≥ rlist.
- Алогритм минимизации энергии.
- Модель, которой описывался растворитель
implicit_solvent = No
Растворителя не было. - Утряска растворителя (файл pr.mdp):
- Для биополимеров, укажите параметр который обуславливает неподвижность биополимера:
define = -DPOSRES
Включает файл posre.itp с номерами неподвижных атомов в топологию. - Число шагов: 10^4.
- Длина шага: 1 фс.
- Алгоритм расчёта электростатики и Ван-дер-Ваальсовых взаимодействий.
coulombtype = pme
Быстрое вычисление электростатики по методу Эвальда Particle-Mesh.vdw-type = Cut-off
- Алгоритмы термостата и баростата.
Tcoupl = Berendsen
Контроль температуры термостатом Берендсена.Pcoupl = no
Без контроля давления. Это означает фиксированный размер ячейки.
- Для биополимеров, укажите параметр который обуславливает неподвижность биополимера:
- Основной расчёт МД (файл md.mdp):
- Время моделирования: 7 часов 12 минут 42 секунды
количество процессоров: 16
эффективность масштабирования: 100%. - Длина траектории: 20 нс.
- Число шагов: 10^7.
- Длина шага: 2 фс.
- Алгоритм интегратора: md,
Leap-frog алгоритм для интегрирования Ньютоновских уравнений движения. - Алгоритм расчёта электростатики и Ван-дер-Ваальсовых взаимодействий.
coulombtype = pme vdw-type = Cut-off
- Алгоритмы термостата и баростата.
Tcoupl = v-rescale
Контроль температуры по методу "Velocity rescale".Pcoupl = Berendsen
Контроль давления с помощью экспоненциальной релаксации. Ячейка пересчитывается каждый шаг.
- Время моделирования: 7 часов 12 минут 42 секунды
Анализ результатов
- Начнём с визуального анализа движений молекул. Переводим траекторию
в формат PDB и смотрим в PyMOL:
trjconv -f pep_md.xtc -s pep_md.tpr -o pep_pbc_1.pdb -skip 20 -pbc mol trjconv -f pep_md.xtc -s pep_md.tpr -o pep_fit_1.pdb -skip 20 -fit rot+trans
Сохранённый роликУже на втором кадре происходит изменение структуры, это модель 1, 200 фемтосекунд от начала. Видно, как в основном C-конец альфа-спирали то расплетается, то принимает прежнее положение. На кадре 33 видно наиболее сильное изменение структуры, оба конца расплелись. Но есть спираль ещё не расплавилась к конце моделирования. Видимо, траектория оказалась недостаточной.
- Определим средне-квадратичное отколнение в ходе моделирования. Так как у нас происходит конформационный переход сначала расчитаем отклонение в ходе все симуляции относительно стартовой структуры.
g_rms -f pep_md.xtc -s pep_md.tpr -o rms_1
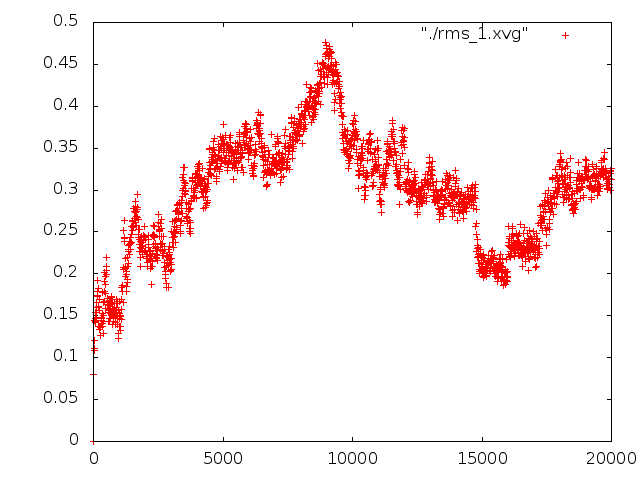
Заметен сильный рост средне-квадратичного отколнения вначале, то есть молекула сразу уходит из своего начального состояния. А в ходе моделирования отклонение то растёт, то убывает, не опускаясь до нуля. Это говорит о том, что в формамиде начальная эталонная альфа-спираль не оптимальна.
Также видно, что RMSD не выходит за пределы 0.5, что довольно мало. Это говорит о том, что пептид несильно меняет свою конформацию, сохраняя в целом альфа-спиральную структуру.И относительно каждой предыдущей структуры на растоянии 400 кадров. Если ближе к концу закончился конформационный переход, то отколнение должно уменьшаться.
g_rms -f pep_md.xtc -s pep_md.tpr -o rms_2 -prev 400
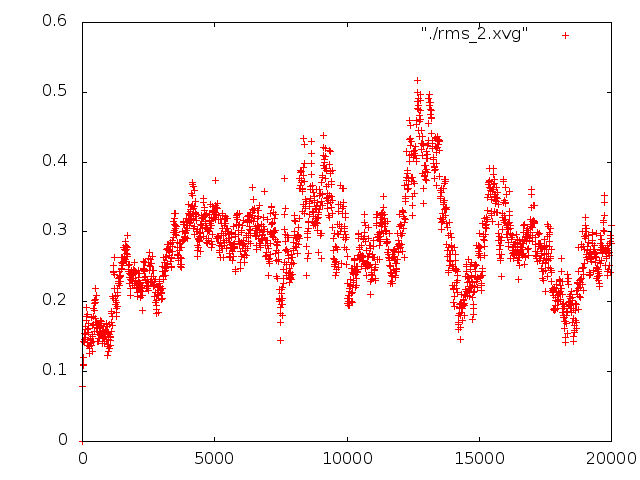
К концу моделирования данное отклонение не уменьшается, то есть молекула не принимает какую-либо устойчивую конформацию.
- Определим изменение гидрофобной и гидрофильной поверхности в ходе конформационного перехода.
g_sas -f pep_md.xtc -s pep_md.tpr -o sas_pep.xvg
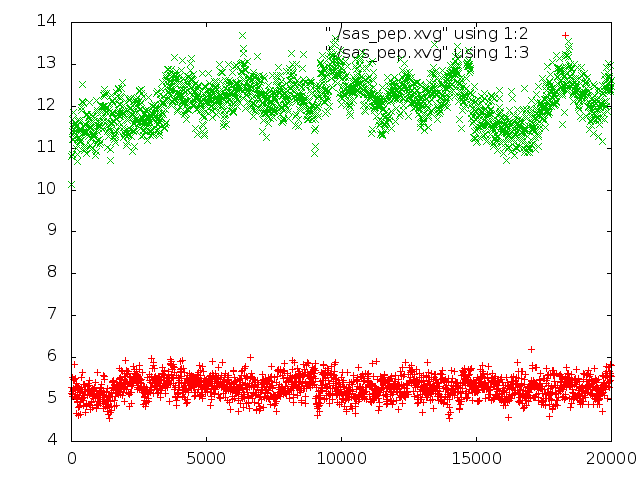
Красным цветом обозначены значения для гидрофобной поверхности, зелёным - для гидрофильной.
Гидрофобная поверхность практически не изменяется. Она включает в себя только гидрофобные остатки. Как они были направлены от спирали в раствор вначале, так и сохранили своё положение до конца. А гидрофильная поверхность слегка изменялась, это отражает "расплетание" и "заплетание" конца спирали.
- Традиционным анализом является расчёт колчества образуемых водородных связей. Если мы будем исследовать связи между пептидом и пептидом, то это будут водородные связи в пептиде.
g_hbond -f pep_md.xtc -s pep_md.tpr -num hbond_pep
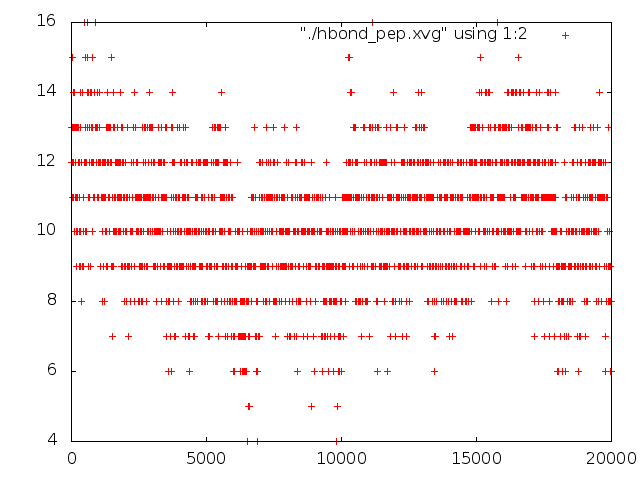
Видно, что количество водородных связей внутри пептида постоянно меняется, опускаясь даже до 4, хотя возможно как минимум 16. Значение 4, вероятно, соответсвует кадру 33, где спираль наиболее сильно расплелась.
Не менее интересно будет изучить количество вдородных связей пептид-формамид.
g_hbond -f pep_md.xtc -s pep_md.tpr -num hbond_pep_sl
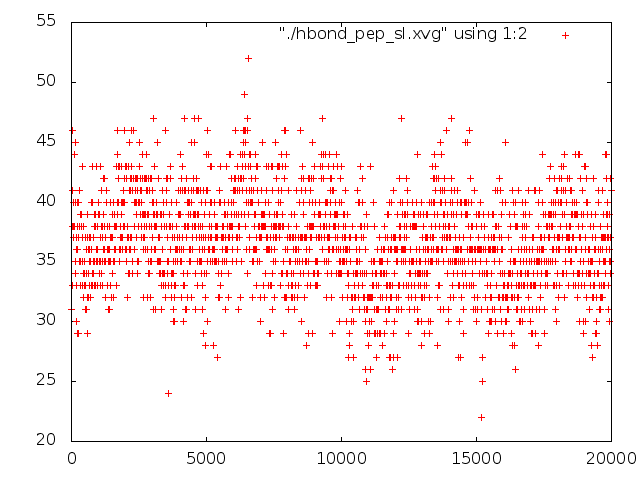
Количество водордных связей с растворителем то растёт, то убывает и не приходит к какому-то определённому значению.
- Если у нас происходит разрушение вторичной структуры, то надо построить зависимость вторичной структуры от времени:
do_dssp -f pep_md.xtc -s pep_md.tpr -o ss # Для просмотра переведём xpm в eps xpm2ps -f ss.xpm -o ss.eps -by 10 # Переведём в картинку convert ss.eps ss.png
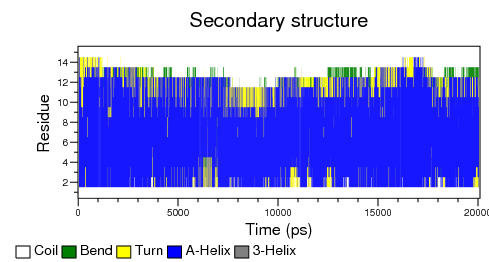
Расчёт вторичной структуры совпадает со всеми наблюдениями. Мы видим, что более активно она меняется на C-конце, чем на N-конце.