На главную страницу четвертого семестра
|
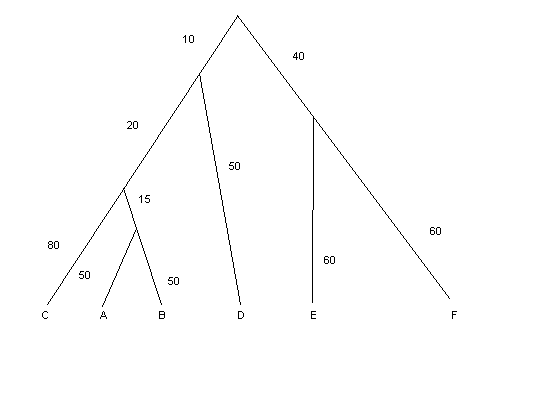
| Цифрами обозначены соответствующие эволюционные расстояния, буквами - потомки (листья). Эволюционные расстояния расчитывалисть как число мутаций на 100 нуклеотидов. Это дерево укорененное, не ультраметрическое, т.к. расстояния от корня до потомков не везде одинаковые. |
Далее это дерево было представлено, как множество разбиений листьев:
A B C D E F
. . * * * *
. . . * * *
* * * * . .
| Точками обозначаются те листья, чья общая ветвь "отделяется" от всех остальных, помеченных звёздочками. Ветви, несущие единственный лист, не были обозначены ввиду своей неинформативности. |
| Outfile | n | Script |
| ABCD | 151 | msbar AMPA_ECOLI_gene.fasta ABCD -point 4 -count 151 -auto |
| ABC | 302 | msbar ABCD ABC -point 4 -count 302 -auto |
| AB | 227 | msbar ABC AB -point 4 -count 227 -auto |
| A | 756 | msbar AB A -point 4 -count 756 -auto |
| B | 756 | msbar AB B -point 4 -count 756 -auto |
| C | 1209 | msbar ABC C -point 4 -count 1209 -auto |
| D | 756 | msbar ABCD D -point 4 -count 756 -auto |
| EF | 605 | msbar AMPA_ECOLI_gene.fasta EF -point 4 -count 605 -auto |
| E | 907 | msbar EF E -point 4 -count 907 -auto |
| F | 907 | msbar EF F -point 4 -count 907 -auto |
| Алгоритм | Дерево | Ветви |
| Исходное | 
| A B C D E F . . * * * * + . . . * * * * * * * . . + |
| Наибольшее правдоподобие | +---B | | +-------------F | +-------------4 | +---3 +-------------E | | | 1---2 +-----------D | | | +------------------C | +-----------A | A B C D E F . . * * * * + . . . * * * * * * * . . + |
| Neighbor-joining | +--B ! ! +------------------C 3---4 ! ! +------------D ! +---2 ! ! +-------------E ! +--------------1 ! +--------------F ! +-----------A | A B C D E F . . * * * * + . . . * * * * * * * . . + |
| UPGMA |
+--------------A
+------------1
+---2 +--------------B
! !
+-------------4 +---------------------------D
! !
--5 +-------------------------------C
!
! +----------------------------E
+----------------3
+----------------------------F
| A B C D E F . . * * * * + . . * . * * * * * * . . + |