Исследование структуры тРНК
В файле приведены координаты атомов следующих молекул: 1.TRANSFER RIBONUCLEIC ACID (YEAST, PHE)(цепи D, E, F).Синтезирована 2.MOLECULE OF ELONGATION FACTOR TU (EF-TU)(цепи A, B, C).Взята из организма THERMUS AQUATICUS Для исследования была выбрана цепь F тРНК со следующей последовательностью:
[1] 5' - GCGGAUUUAGCUCAGUUGGGAGAGCGCCAGACUGAAGAUCUGGAGGUCCUGUGUUCGAUCCACAGAAUUCGCACCA - 3' [76]
где 1 и 76 - номера первого и последнего нуклеотида.В данной последовательности на 3'-конце имеется триплет CCA, к которому присоединяется аминокислота. В PDB приведены координаты его атомов.
По имеющимся в файле данным была определена длина стеблей РНК: 1.акцепторный стебель:1-7 нуклеотиды и комплементарные ему 66-72 2.Т-стебель: состоит из 49-53 и комплементарных ему 61-65 3.D-стебель: 10-13 и 22-25 4.антикодоновый стебель: 39-43 и 27-31 5.антикодон: 34-36
| Скрипт для получения изображения | |
 |
restrict none select *:F color white backbone 80 define act 1-7:F or 66-72:F select act color red pause define dst 10-13:F or 22-25:F select dst color blue pause define tst 49-53:F or 61-65:F select tst color green pause define anti 27-31:F or 39-43:F select anti color orange select 34-36:F cpk 100 wireframe 50 color purple |
Структуру стеблевых дуплексов поддерживают 20 канонических и 11 неканонических пар оснований.
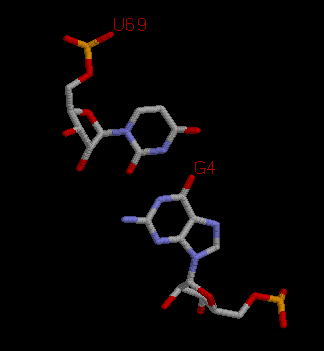 |
Примером неканонического взаимодействия может служить взаимодействие 4G и 69U нуклеотидов. |
Вариабельная петля в данной структуре отсутствует В Т-петле отсутствует остаток тимидина Дигидроуридины в D-петле отсутствуют
 |
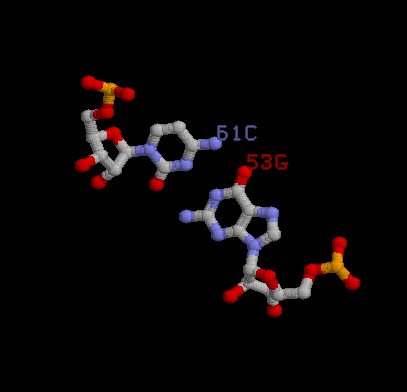
Была исследована возможность стэкинг-взаимодействия
между основаниями конца акцепторного стебля (№66-68) и начала Т-стебля (№63-65). В пользу стекинг-взаимодействия свидетельствуют то, что по данным файла 1TTT_old.out площадьперекрывания в выбранной паре №72 GU/AC равна 9.46, тогда как максимальный показательпо сравнению с другими парами равен 15.68, что достаточно много для того, чтобы с высокой долей вероятности предположить в данном месте стэкинг-взаимодействие. На данной картинке изображено наложение оснований исследуемой пары. |
 |
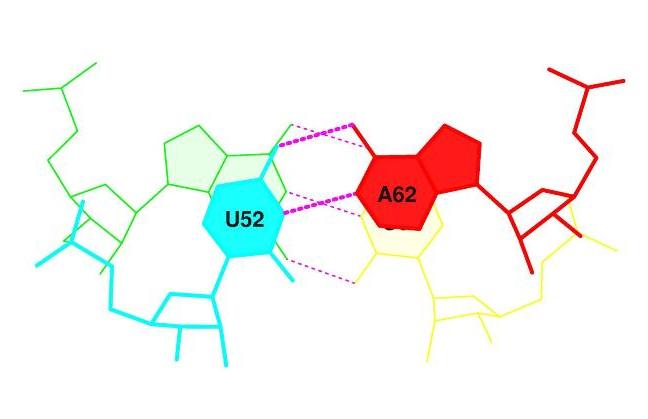
Дополнительные водородные связи присутствуют между
основаниями Т-петли. Это каноническое взаимодействие между 207(53) и 215(61) нуклеотидами, а также взаимодействие между модифицироваными нуклеотидами 54_:[5MU]u-**-xa[1MA]:_58. Взаимодействие между нуклеотидами последней пары представлены на следующей далее картинке: |
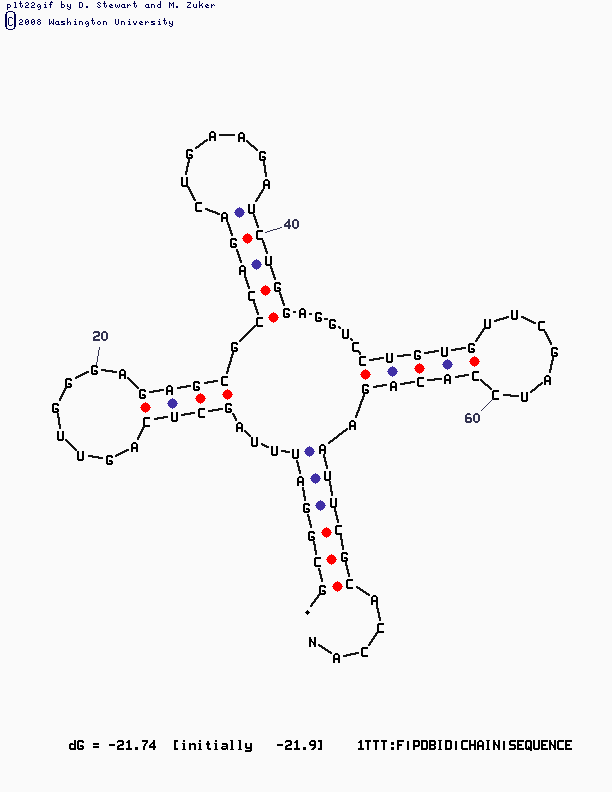
Реальная и предсказанная вторичная структура тРНК из файла 1TTT.pdb
| Участок структуры | Позиции в структуре (по результатам find_pair) |
Результаты предсказания с помощью einverted |
Результаты предсказания по алгоритму Зукера |
| Акцепторный стебель | 5' 1-7 3' 5' 66-72 3' Всего 7 пар |
не предсказано | предсказано 6 пар из 7 |
| D-стебель | 5' 10-13 3' 3' 22-25 5' Всего 4 пары |
5' 12-16 3' 3' 31-35 5' 4 пары |
все 4 пары предсказаны верно |
| T-стебель | 5'49-53 3' 3' 61-65 5' 5 пар |
5' 49-53 3' 3'61-65 5' 5 пар |
все пары предсказаны верно |
| Антикодоновый стебель | 5' 27-31 3' 3' 39-43 5' 5 пар |
не предсказано | все пары предсказаны верно |
| Общее число канонических пар нуклеотидов | 21 | 9 | 20 |
Программа einverted
Программа einverted используется для нахождения инвертированных участков в нуклеотидных последовательностях. Программе был подан файл с последовательностью, при этом сохранялись все настройки программы за исключением параметра Score, от величины которого зависел получаемый результат. На выходе были получены 2 файла: sequence.fasta и sequence.inv:
SEQUENCE: Score 15: 5/5 (100%) matches, 0 gaps
12 tcagt 16
|||||
35 agtca 31
SEQUENCE: Score 15: 5/5 (100%) matches, 0 gaps
49 ctgtg 53
|||||
65 gacac 61
По результатам, выданным программой можно сделать вывод, что она не является лучшей для предсказания вторичной структуры, так как была способна обнаружить лишь 2 участка из 4-ёх, что, вероятно, связано со способностью программы обнаруживать лишь канонические взаимодействия.
 |
При использовании программы mfold, необходимой для
предсказания вторичной структуры тРНК по алгоритму Зукера, на вход был подан файл tRNA2.fasta, с находящейся внутри нуклеотидной последовательностью выбранной произвольным образом цепи F РНК. Была осуществлена команда mfold SEQ=tRNA2.fasta. Затем команда вводилась с добавлением параметра P со значениями 10, 15, 20, 25. В большей мере полученные результаты были неудовлетворительными, и лишь при значении P=10 приемлемыми. В результате этих действий и было получено данное изображение.и |
По результатам, полученным в результате использования программ, можно сделать вывод, что программа einverted не слишком хороша для решения поставленной
задачи, так как с её помощью правильно были определены лишь D- и T-стебель, в то время как акцепторный и антикодоновый стебли не были определены вовсе.
Что же касается программы mfold, то она определила вторичную структуру достаточно точно: лишь в акцепторном стебле была потеряна одна пара, всё остальное
было предсказано верно. То есть можно сделать вывод, что программа mfold вполне может использоваться для предсказанная вторичная структура тРНК с достаточной точностью.
©Черниогло Елена