Множественные выравнивания
Малые дельта-антигены
Сначала было получено выравнивание вирусных белков, называемых "малыми дельта-антигенами". Если сравнвать выравнивание с невыравненным набором последовательностей, то видно, что после первой же мутации последовательности начинают "плыть" (гомологичные фрагменты оказываются не друг под другом)Если же смотреть на выравненные последовательности, то видны большие повторяющиеся блоки.
Набор гомологов моего белка
Мной было построено выравнивание моего белка с 10 гомологами. Выравнивание представляет собой набор "островков" с повышенной долей консервативных позиций, между которыми располагается примерно такое же количество плохо выравниваемых а.о. По-видимому консервативность наблюдается там, где а.о. образуют активный центр данного фермента.Например, с 83-й по 108-ю позиции выравнивания (с 74 по 99 а.о. в моем белке THIE_BACSU) расположен фрагмент из двух таких "островков". Каждый из них - это β-тяж, участвующий в образовании активного центра белка, а невыравненный промежуток между ними - это α-спираль, соединяющие эти два тяжа (что, на мой взгляд, странно, т.к. до этого нам объясняли, что изменения в а.о. спиралей и слоев редки, т.к. они изменяют структуру довольно существенно. А мутации в данной спирали самые разнообразные, не всегда проходящие с сохранением свойств а.о. Хорошо хоть число а.о. в спирали остается постоянным)
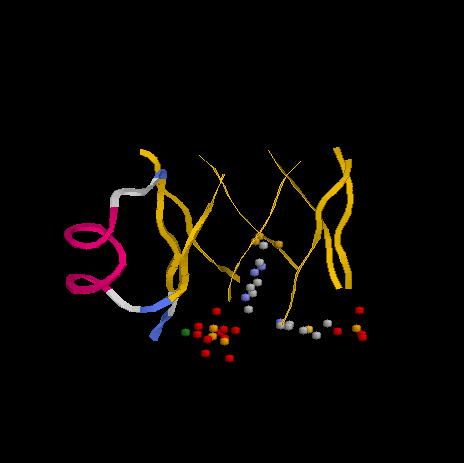
Слева - выступает α-спираль, соединяющая два из β-тяжей, образующих колодец - активный центр фермента. Шариками обозначены атомы лигандов.
Неправдопобными, на мой взгляд, кажутся выравнивания одной или совсем небольшого количества а.о. ("немного" - это, конечно, не точный научный термин, но интуитивно, надеюсь, понятно, что имеется в виду). Примерами таких не заслуживающих доверия фрагментов могут быть части выравнивания со 165 по 172 а.о. (нумерация по выравниванию), 222 а.о. (просто случайно выравнялось, по-моему).
Другие программы для выравнивания
Программы mafft и edialign, в общем, выдают такие же выравнивания в тех местах, где существуют "островки" сходства, и расходятся во мнении там, где выравнивания единичны и, скорее всего, никакого биологического смысла не несут. Кстати, это отличный способ искать гомологичные фрагменты - пропускать набор последовательностей через несколько разных программ и смотреть, где они сошлись во мнении :)Edialign, помимо выравнивания, выдает общую информацию о сравниваемых последовательностях и (отличительная особенность!) дерево, полученное в ходе выравнивания.
Выравнивание, полученное программой Mafft (.msf)
Выравнивание, полученное программой Edialign (.msf)
Программы обработки выравниваний
Программа Consambig
Программа Consambig строит маску всех последовательностей в выравнивании. Формат - fasta. Строчные буквы используются, чтобы показать маску в тех частях выравнивания, где имеются гэпы и, соответственно, выравниваются не все последовательности.Программа DistMat
Программа DistMat подсчитывает эволюционное расстояние между последовательностями (расстояние по смыслу обратно весу выравнивания - чем меньше расстояние, тем в более тесных родственных связях находятся белки. Например, наименьшее расстояние было показано для белков из бактерий одного класса - Bacillus)Программа Plotcon
Программа Plotcon строит график распределения сходства а.о. по выравниванию.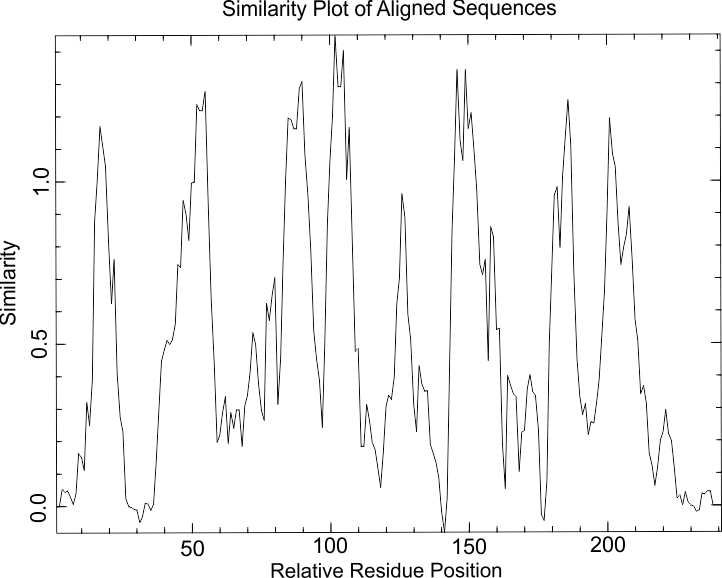
График распределения сходства на выравненных последовательностях моего белка и его гомологов. Имеются четко выраженные пики распределений, соответствующие консервативным фрагментам в выравнивании.
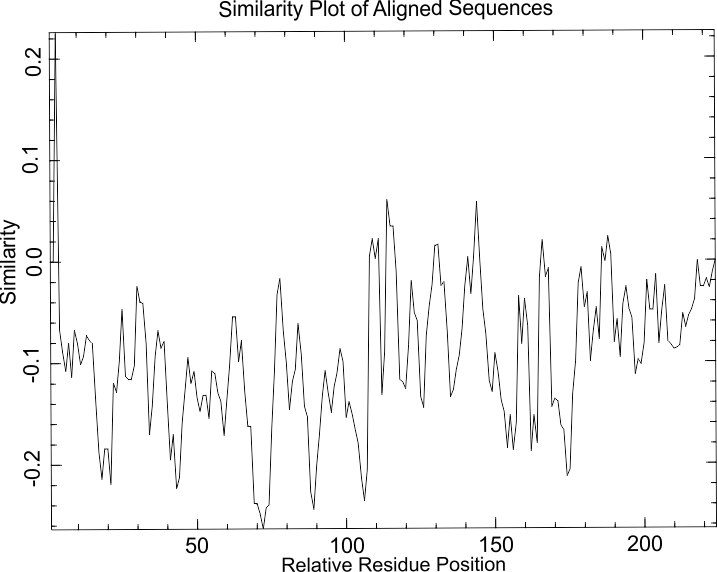
График распределения сходства на невыравненных последовательностях моего белка и его гомологов. Обратите внимание - масштаб по шкале "Сходство" другой, теперь все значения колеблются около ноля. В самом начале - значения выше, т.к. первый а.о. у всех белков - метионин, по нему все гомологи сходны.