Занятие 9.
Цель данного занятия ознакомится с возможностями гомологичного моделирования комплекса белка с лигандом. В этом занятии мы будем пользоваться пакетом Modeller. Это программное обеспечение распространяется бесплатно для академических пользователей.Мы будем работать с белком лизоцимом из указанного организма. Используя известную структуру лизоцима форели как образец, нам необходимо построить модель комплекса белка с лигандом.
1.Постройте выравнивание последовательности из структуры ID: 1lmp (1lmp) и предложенного белка (lysc_erypa). Рекомендуемые ресурсы и программы: Clustal и GeneDoc Полученное выравнивание сохраните в формате PIR (в txt align.pir.txt).
2.Модификация файла выравнивания:
Переименуйте последовательность в файле выравнивания как в примере:
Было Стало >P1;uniprot|P37712|LYSC_CAMDR >P1;seq >P1;1LMP__|PDBID|CHAIN|SEQUENCE >P1;1lmpПосле имени последовательности моделируемого белка добавьте строчку:
sequence:ХХХХХ::::::: 0.00: 0.00эта строчка описывает входные параметры последовательности для modeller. После имени последовательности белка-образца добавьте:
structureX:1lmp_now.ent:1 :A: 132 :A:undefined:undefined:-1.00:-1.00эта строчка описывает, какой файл содержит структуру белка с этой последовательностью, номера первой и последней аминокислот в структуре, идентификатор цепи и т.д. В конце каждой последовательности добавьте символы
/.Символ "/" означает конец цепи белка. Точка указывает на то, что имеется один лиганд (если бы было два лиганда стояли бы две точки).
3.Модификация файла со структурой: удалите всю воду из структуры (в текстовом редакторе) всем атомам лиганда присвойте один и тот же номер "остатка" (MODELLER считает, что один лиганд = один остаток) и модифицируйте имена атомов каждого остатка, добавив в конец буквы A, B, C. Смысл операции в том, что атомы остатка 130 имели индекс А, атомы остатка 131 имели индекс В и т.д. . После модификации имен атомов измените номера остатков на 130. Пример: Было Стало
HETATM 1014 O7 NAG 130 HETATM 1014 O7A NAG 130 HETATM 1015 C1 NAG 131 HETATM 1015 C1B NAG 130сохраните в файле 1lmp_now.ent
4.Создание управляющего скрипта lysc_erypa.py
from modeller.automodel import *
class mymodel(automodel):
def special_restraints(self, aln):
rsr = self.restraints
for ids in (('OD2:120:A', 'O6A:149:B'),
('NE1:82:A', 'O3B:149:B'),
('OD2:71:A', 'O1LC:149:B')):
atoms = [self.atoms[i] for i in ids]
rsr.add(forms.upper_bound(group=physical.upper_distance, feature=features.distance(*atoms), mean=3.5, stdev=0.1))
env = environ()
env.io.hetatm = True
a = mymodel(env, alnfile='align.pir', knowns=('1lmp'), sequence='seq')
a.starting_model = 1
a.ending_model = 5
a.make()
В скрипте указано:
что нужно использовать стандартные валентные углы в полипептидной цепи (строчка 4)
что дополнительно нужно сохранять взаимное расположение определенных пар атомов (3.5 ангстрема);
В данном случае трех атомов белка, образующих водородные связи с тремя атомами лиганда - строчки 5-7 с ID пар атомов; параметры взаимного расположения атомов пары описаны в строчке 9-10. 3 точки могут однозначно расположить сложную структуру в пространстве, поэтому мы выбираем водородные связи как источник данных точек.
что ковалентные связи в гетероатомах нужно вычислять по расстояниям между атомами (так же, как это делает Rasmol), строчка 12
что имя файла с выравниванием и имена последовательностей образца и моделируемого белка, строчка 13 (а имя файла со структурой содержится в выравнивании)
что число и номера моделей, которые нужно построить (в данном примере 5 моделей), строки 14-15
что пора строить модель строчка 16
5.Запустите исполнение скрипта командой
mod9v7 myscript &
6. Получили пять похожих результатов. У всех у них хвост белка lysc_erypa торчит наружу. Т.к. для него нет гомологичных остатков в образце. В выравнивании 18 аминокислотам на С-конце нет гомологичных аминокислот в последовательности белка-образца.
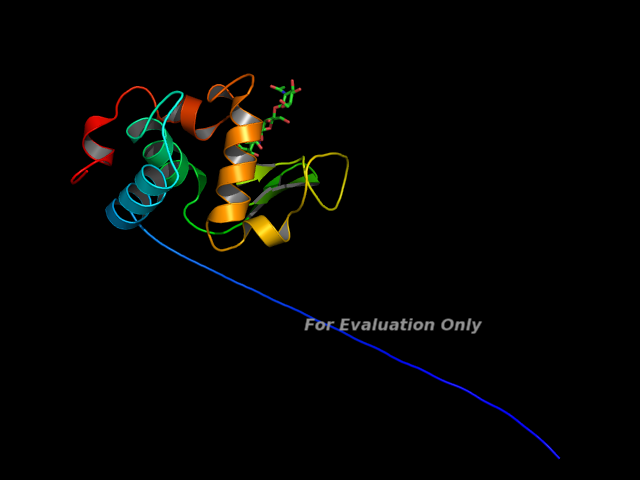
Проверим качество модели с помощью WHATIF: 1)
Structure Z-scores, positive is better than average: 1st generation packing quality : -2.797 2nd generation packing quality : -3.058 (poor) Ramachandran plot appearance : 0.629 chi-1/chi-2 rotamer normality : -1.361 Backbone conformation : -0.670 RMS Z-scores, should be close to 1.0: Bond lengths : 0.935 Bond angles : 1.309 Omega angle restraints : 0.766 Side chain planarity : 0.296 (tight) Improper dihedral distribution : 1.010 Inside/Outside distribution : 1.0422)
Structure Z-scores, positive is better than average: 1st generation packing quality : -2.750 2nd generation packing quality : -2.850 Ramachandran plot appearance : 0.016 chi-1/chi-2 rotamer normality : -2.339 Backbone conformation : -1.116 RMS Z-scores, should be close to 1.0: Bond lengths : 0.927 Bond angles : 1.280 Omega angle restraints : 0.773 Side chain planarity : 0.366 (tight) Improper dihedral distribution : 1.102 Inside/Outside distribution : 1.045 ==============3)
Structure Z-scores, positive is better than average: 1st generation packing quality : -2.930 2nd generation packing quality : -3.155 (poor) Ramachandran plot appearance : -0.365 chi-1/chi-2 rotamer normality : -2.240 Backbone conformation : -1.033 RMS Z-scores, should be close to 1.0: Bond lengths : 0.928 Bond angles : 1.322 Omega angle restraints : 0.856 Side chain planarity : 0.364 (tight) Improper dihedral distribution : 1.021 Inside/Outside distribution : 1.054 ==============4)
Structure Z-scores, positive is better than average: 1st generation packing quality : -2.872 2nd generation packing quality : -2.710 Ramachandran plot appearance : 0.224 chi-1/chi-2 rotamer normality : -2.380 Backbone conformation : -0.991 RMS Z-scores, should be close to 1.0: Bond lengths : 0.935 Bond angles : 1.304 Omega angle restraints : 0.802 Side chain planarity : 0.347 (tight) Improper dihedral distribution : 1.124 Inside/Outside distribution : 1.047 ==============5)
Structure Z-scores, positive is better than average: 1st generation packing quality : -2.733 2nd generation packing quality : -2.547 Ramachandran plot appearance : -0.567 chi-1/chi-2 rotamer normality : -1.777 Backbone conformation : -0.733 RMS Z-scores, should be close to 1.0: Bond lengths : 0.937 Bond angles : 1.324 Omega angle restraints : 0.871 Side chain planarity : 0.262 (tight) Improper dihedral distribution : 1.025 Inside/Outside distribution : 1.041 ==============
Выберем несколько параметров для сравнения:
1) 2) 3) 4) 5)
Ramachandran plot appearance 0.629 0.016 -0.365 0.224 -0.567
chi-1/chi-2 rotamer normality -1.361 -2.339 -2.240 -2.380 -1.777
Backbone conformation -0.670 -1.116 -1.033 -0.991 -0.733
Side chain planarity 0.296 0.366 0.364 0.347 0.262
Improper dihedral distribution 1.010 1.102 1.021 1.124 1.025
Из таблицы видно, что самая лучшая структура первая.